CE | MDD 의료기기 / 유럽 제품인증 소개
의료기기 지침은 유럽 연합 내에서 의료기기에 관한 법률을 조화하기 위한 것입니다. 법적으로 유럽 시장에 의료기기들을 판매하기 위해서는 제조자들은 MD지침(Medical Device Directive)의 요구 사항을 준수해야 합니다. 신청자의 제품 및 품질 시스템을 평가해야 하며 제조자는 제품들을 판매 하기전에 CE 마크를 부착해야 합니다.
적용 품목
- 의료기기 지침은 '의료기기'의 정의에 따르는 장치에 적용 가능합니다. 의료기기란 제조자의 의도대로 인간에게 사용되는 목적을 가진 소프트웨어를 포함하는 단독 또는 조합으로 사용되는 기계, 장치, 기기, 재료 또는 기타 물품입니다.
- 진단, 예방, 감시, 치료 또는 질병의 경감, 진단, 감시, 치료, 상해 또는 장애에 대한 완화 또는 보상, 조사, 교체 또는 해부학적 또는 생리학적 과정의 수정, 개념의 제어, 약리적, 면역적 또는 신진대사적 수단에 의해 인체 내에 또는 인체상에 의도한 주요 작용을 달성하지는 않지만 그런 수단에 의해 그 기능을 도와줄 수 있는 것을 말합니다.
❉ ❉ ❉
적격성 평가 절차
❀ 분류법
- 의료기기 지침서(93/42/EEC)의 부속서 IX는 의료기기를 분류할 수 있는 18개의 규칙을 제공합니다. 이 규칙들에 따라 의료기기는 의료기기의 의도된 목적에 따라 분류됩니다.
✧ Rule 1 ~ 4 : Non-invasive 기기
✧ Rule 5 ~ 8 : Invasive 기기
✧ Rule 9 ~ 12 : 능동형 기기
✧ Rule 13 ~ 18 : 특별법
- 환자 및 사용자에 대한 의료기기의 위험 평가는 위의 18개의 규칙에 따라 식별되고 분류됩니다. 식별된 위험 값을 바탕으로 적격성 평가 절차가 결정됩니다. 위험 값이 높을수록 적격성 평가 요구 사항이 엄격 해집니다.
- Class I 의료기기는 부속서 VII의 평가 절차를 따릅니다.
❀ Class I의 절차 (측정 기능 포함)
제조사의 선택 가능 사항 :
• 부속서 VII 섹션 3 및 부속서 IV에 따른 각각 또는 통계적으로 선택된 샘플에 대한 도량형에 관한 요구사항을 만족하는 적격성의 검증 및 기술 문서화
• 부속서 VII 섹션 3 및 통계학적 요구 사항과 연관된 부속서 V에 의한 제품 품질 시스템의 평가 및 기술 문서화
• 부속서 VII 섹션 3 및 통계학적 요구 사항과 연관된 부속서 VI에 의한 의료기기의 품질 평가 및 기술 문서화
❀ Class I의 절차 (멸균 포함)
제조사의 선택 가능 사항 :
• 부속서 VII 섹션 3 및 멸균 상태와 연관된 부속서 V에 의한 제품 품질 시스템의 평가 및 기술 문서화
❀ Class IIa 의료기기 절차
제조사의 선택 가능 사항 :
• 부속서 VII 섹션 3 및 부속서 IV의 섹션 8에 의한 각각 또는 통계학적으로 선택된 샘플에 대한 적격성 검증 및 기술 문서화
• 부속서 VII 섹션 3 및 부속서 V의 섹션 6(제조사의 현장에서 심사 된)에 의한 제품 품질 시스템의 평가 및 기술 문서화
• 부속서 VII 섹션 3 및 부속서 VI의 섹션 6(제조사의 현장에서 심사 된)에 의한 의료기기 품질의 평가 및 기술 문서화
• 부속서 VII 섹션 3 및 부속서 II(섹션 4에 따라 개발 평가가 제외 된)에 의한 전체적인 품질 시스템이 제조자의 현장에서 심사 된 평가 및 기술 문서화
❀ Class IIb 의료기기 절차
제조사의 선택 가능 사항 :
• 부속서 III 섹션 3 및 제조된 각각에 대한 적격성 검증 및 유형 검사 와 기술 문서화 : 부속서 IV에 의해 통계학적으로(무작위로) 선택 된 샘플이나 섹션 5에 의해 검증 되어야 합니다.
• 부속서 III 섹션 3 및 부속서 V(제조사의 현장에서 심사 된)에 의해 제조 품질 시스템의 평가 및 유형 검사와 기술 문서화
• 부속서 III 섹션 3 및 부속서 VI(제조사의 현장에서 심사 된) 섹션 6에 의해 의료기기 품질의 평가 및 유형 검사의 기술 문서화
• 부속서 II 섹션 3.2 및 부속서 II(섹션 4에 의해 개발 평가가 제외된)에 의해 제조사의 현장에서 심사가 이루어진 모든 품질 시스템의 평가에 대한 기술 문서화
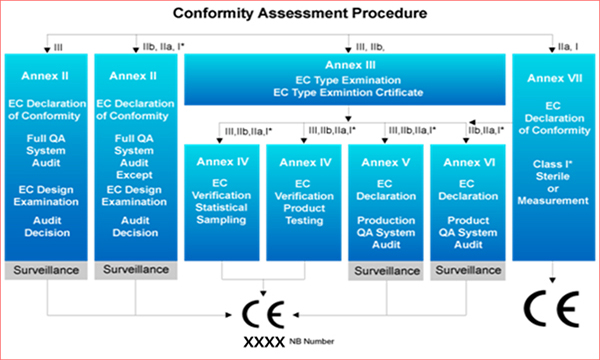 적합성 평가 절차
적합성 평가 절차
❀ Class III 의료기기 절차
제조사의 선택 가능 사항 :
• 부속서 II 섹션 3.2 및 부속서 II(제조사의 현장에서 심사된)에 의한 모든 품질 시스템의 평가 및 섹션 4.2에 의해 개발의 설명에 대한 기술 문서화
• 부속서 III 섹션 3 및 제조된 각각에 대한 적격성 검증 및 유형 검사의 기술문서화 : 부속서 IV 섹션 6에 의해 통계학적으로(무작위) 선택된 샘플에 대한 섹션5에 의해 검증이 이루어져야 합니다.
• 부속서 III 섹션 3 및 부속서 V 섹션 3.2에 의해 제품 품질 시스템(제조사의 현장에서 심사된)의 평가 및 유형 검사의 기술 문서화
❉ ❉ ❉
MDD 종료 시점
- 유럽의 의료기기 지침인 MDD (93/42/EEC)는 2020년 5월 26일부터 MDR로 완전히 대체됩니다. 이후에는 MDD로의 CE인증 신청이 불가능하며, 새로운 의료기기는 MDR (2017/745/EU) 요구사항을 충족시켜야만 합니다.
- 하지만 강제적용 시점 전에 MDD의 적합성 평가를 마무리하여 인증서를 발급받은 경우, 최대 2024년 05월 27일까지 인증서의 효력을 인정받아 제품을 유럽 관할구역내에 출하할 수 있습니다.
- 유럽의 Competent Authority에서 공식적으로 발행한 상기의 자료를 확인해 보실 수 있습니다.
❉ ❉ ❉
CE / MDD 제공 서비스
- IGC는 현재 3개의 Notified Body(이하 NB)기관과 협력하여 CE/MDD 프로젝트를 진행하고 있습니다.
- 이로 인해, Class l 부터 Class lll까지 거의 모든 스콥에 관하여 예외 없이 신청을 진행할 수 있습니다.
- 수 많은 프로젝트를 진행하며 쌓은 경험을 토대로 귀사의 CE/MDD 인증 획득을 위하여 지원을 아끼지 않겠습니다.
❉ ❉ ❉
우리가 제공하는 관련 서비스
01경영시스템 인증
02제품인증 서비스
03제품 시험
04심사자격 인증 서비스
05심사원 교육 및 양성
CE MDR | 의료기기 / 유럽 제품인증 소개
CE Marking 이란 ?
의료기기 규제는 유럽 연합 내에서 의료기기에 관한 법률을 조화하기 위한 것입니다. 법적으로 유럽 시장에 의료기기들을 판매하기 위해서는 제조자들은 MDR(Medical Device Regulation)의 요구 사항을 2020년 강제적용 이후 준수해야 합니다. 신청자의 제품 및 품질 시스템을 평가해야 하며 제조자는 제품들을 판매 하기전에 CE 마크를 부착해야 합니다.
MDR 개요
- 적용규격 : Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 Ap
- 법규내용 : Council Directive 93/42/EEC (MDD) + Council Directive 90/385/EEC (AIMD)
- 발효일자 : 2017년 05월 25일
- 강제적용일자 : 2020년 05월 26일, MDR 발효 후, 3년의 유예기간
- 목적 : 유럽 내 좀 더 안전하고, 효과적이고, 혁신적인 의료기기 시장을 만드는 것
- MDD 인증서 최종 유효일자 : 2024년 05월 26일 (판매는 2025년 05월 27일까지)
- 지침(Directive)에서 규제(Regulation)로 바뀜. Directive보다 Regulation 은 변경이 더 자주 일어나며, 강제성이 강화됨.
주요 개정 내용
- 심사기관 (NB)에 대한 보건당국의 강력한 통제 : Sub-contractor 관리강화
- Harmonized Standard 외에 제품별 공통기술사양(Common Specification) 도입 및 적용
- 경제운영자 [Manufacturer, Importer, Distributor, Authorized Representative]의 책임 강화
- EUDAMED 시스템에 등록 의무화(제조자와 경제운영자간의 표준계약서 필요)
- 제조업체에 대한 NB의 강력한 통제
- Pre/Post Market 임상평가의 확장과 모든 임상 조사에 대한 관리, 제조자의 시판 후 시장감시(Vigilance & PMS) 강화
- EUDAMED(European database on medical devices) 기반의 전산시스템 정착 : 제품등록, UDI, 경제운영자등록, NB 및 인증서, 임상조사, Vigilance & PMS, 시장조사(보건당국)
- UDI System의 개선 및 도입 : ClassⅢ부터 순차 도입. 현재 FDA와 호환가능하나, 변경가능성 있음.
- MDCG(Medical Device Coordination Group) : 보건당국 내 독립적인 전문가들로 구성된 의료기기조정그룹으로 Class III Implantable & Class IIb 능동기기 제품에 대한 임상평가 자문 및 적합성평가 정밀조사, NB평가, Class III 관리
- Class I 재사용수술기구 : Class I 멸균/측정기기처럼 NB 심사대상 → 세척 등의 재가공(Reprocessing) 공정에 대해서만 실시
- 임플란트카드 : 의사, 제조자, 환자, 제조번호, etc.
- 규제 적합성 책임자 (Person Responsible for Regulatory Compliance) : 제조자와 유럽대리인 공통사항으로 1년 이상 품질 & RA 업문 경력, 4년제 대졸이상의 학력을 소지한자로서 기술문서와 제품출시에 책임.
- 의료기기 정의에 속하지 않는 제품들에 대한 MDR 적용 → 필러, 매선, 레이저수술기(제모, 미용목적)
❉ ❉ ❉
TCF(Technical Document)에 포함될 내용
1. MDR Annex1의 GSPR(General Safety and Performance Requirements) Check List 작성
2. Biological Safety Assessment Report (ISO10993-1:2018)
3. Risk Management Plan (ISO14971:2019)
4. Risk Management Report
5. Usability Validation Plan (IEC62366-1:2015)
6. Usability Validation Report
7. Clinical Evaluation Plan (MEDDEV 2.7/1 :2016
8. Clinical Literature Search Protocol
9. Clinical Literature Search Report
10. Adverse Event Search Report
11. Clinical Evaluation Report
12. 공정 (멸균, 세척, 포장, Clean Room) Validation Report 준비
13. Software Validation Plan
14. Software Validation Report
15. 제품도면 및 개략도
16. 제조공정도
17. User Manual (Instructions for use)
18. 사용원자재 List(중요부품) 등
19. Product Label
20. SSCP(Summary of safety and clinical perpormance)매년 up load : Implantable 및 Class III Article 32
21. PSUR(Periodic Safety Update Report) 정기적으로 update : IIa, IIb, III 제품 Article 86
22. DOC (Declaration of Conformity)
❉ ❉ ❉
IGC의 역량 및 관련 제공 서비스
- IGC는 수년간 쌓아온 기술력 및 전문성을 통해 제품 인증의 적합성을 정확하게 평가하여 고객의 지속적인 발전에 일조하고 있습니다.
- IGC는 전세계 주요 시장에서 다양한 특정 범위 및 법적 요구 사항에 대한 최신 지식을 보유하고 있으며, 고객 여러분의 전체 글로벌 운영을 지원하기 위한 지식 및 서비스를 제공하고 있습니다.
- 또한, IGC는 의료기기 분야에서의 다양한 인증 관련 서비스를 제공하고 있습니다.
✦ 93/42/EEC
✦ ISO 13485
❉ ❉ ❉
우리가 제공하는 관련 서비스
01경영시스템 인증
02제품인증 서비스
03제품 시험
04심사자격 인증 서비스
05심사원 교육 및 양성
임상평가 / 유럽 제품인증 소개
임상평가는 의료기기와 관련된 임상 데이터를 수집하고 평가하여 안전성, 효과 및 성능을 검증하고 관련 규제 요건에 대한 적합성을 입증하는 과정입니다.
임상평가는 의료기기의 수명 주기 전체에 걸쳐 실시하는 과정으로, 제조 업체가 연구해야 할 데이터를 승인하기 위해 의료기기를 개발하는 과정 및 기기의 안전성, 효과 및 성능에 관한 새로운 정보를 얻기 위한 과정에서 임상평가를 실시하여야 합니다.
의료기기 생산 기업은 제품의 안전성 및 사용 목적에 따른 효과성을 입증하기 위하여 임상시험을 진행할 때 ISO 14155 규격의 요구사항을 충족하여야 합니다.
MDR과 임상
- MDR(Medical Device Regulation, 유럽 의료기기 규정)이란 유럽시장에 진출하고자 하는 모든 의료기기 제조업체가 준수해야 되는 새로운 의료기기 규정을 말합니다. MDR은 임상과 관련한 데이터 및 평가 과정을 강화하는 프로세스를 요구하며 제조업체는 의료기기의 안전성 및 성능을 입증하기 위해 임상 평가 보고서를 제공하여야 합니다.
- Class III, Class IIb 제품은 임상평가를 필수로 진행하여야 하며, 임상평가 사용 설명서, PMCF 계획을 포함하는 대상이 됩니다.
- 동등한 의료기기의 임상 경험에 대한 데이터 수집, 임상 평가 보고서를 통한 임상 결과 도출, 유사한 기기를 사용한 임상평가를 통해 임상 데이터를 습득할 수 있습니다.
❉ ❉ ❉
임상평가 수행 단계
1. 임상 평가 계획의 수립
2. 임상 데이터 생성 및 식별 (문헌 검색, 임상 경험, 임상 조사)
3. 임상 데이터의 적합성 평가
4. 전체 데이터 분석 및 결론 도출
5. 임상평가 보고서 작성
❉ ❉ ❉
임상시험 자료 필수 사항
-
임상 시험 평가 자료는 다음을 포함하여야 합니다.
✤ 위험평가, 설계된 임상시험에 대한 정당성 평가, 임상 시험단계, 임상시험 브로셔, 임상시험
-
진행보고서, 모니터링 계획, 임상시험 장소 선정 이유, 임상 참가자 동의서, 임상 참가자 식별 방법 등에 대한 항목을 포함하여야 합니다.
✤ 초기 임상 시험 장소에 대한 정보가 있어야 합니다.
✤ 임상 시험 장소에 대한 주기적인 모니터링이 필요합니다.
✤ 부작용이나 기기의 결함 등에 대한 자료를 포함하여야 합니다.
❉ ❉ ❉
PMS란?
- ‘PMS’란 시판 후 감시 시스템이라고도 하며, 제조업체가 EU 시장에 출시한 의료기기에서 얻은 경험을 사전에 수집하고 검토하기 위해 체계적인 절차를 수립하고, 수집된 자료를 최신 상태로 유지하기 위해 제조업체가 수행하는 모든 활동을 의미합니다.
- 제조업체는 각 기기에 대해 위험 등급에 비례하고 기기 유형에 적합한 방식으로 시판 후 감시 시스템을 계획, 수립, 문서화, 구현, 유지 및 업데이트하여야 합니다. 시판 후 감시 시스템은 전체 수명 동안 기기의 품질, 성능 및 안정성에 관한 데이터를 능동적이고 체계적으로 수집, 기록 및 분석하고 필요한 결론을 도출하고 예방 및 시정조치의 결정, 실행 및 모니터링에 적합하여야 합니다.
- MDR에서는 제조업체에 대한 시판 후 요구 사항과 데이터를 사전에 수집하고 이러한 시판 후 활동 및 데이터의 분석을 문서화하고 보고 할 의무가 강화되었습니다.
❉ ❉ ❉
PMS 보고서 작성
- 시판 후 감시 보고서 (Post-Market Surveillance Report, PMSR)
Class I 기기의 제조업체는 시판 후 감시 계획의 결과로 수집된 시판 후 감시 데이터의 분석 결과 및 결론을 요약 한 시판 후 감시 보고서를 준비하여 예방 및 시정에 대한 이론적 근거 및 설명을 작성해야 합니다. 필요한 경우 보고서를 업데이트하고 요청 시 관할 기관에 제공해야합니다.
- 주기적 안전 업데이트 보고서 (Periodic Safety Update Report, PSUR)
Class IIa, Class IIb 및 Class III 기기의 제조업체는 각 기기에 대한 주기적인 안전 관리에 따른 결과 보고서 (PSUR) 및 각 범주 또는 기기 그룹과 관련하여 사후 분석 결과 및 결론을 요약해야 합니다. 시장 감시 데이터는 시판 후 감시 계획의 결과로 취해진 예방 및 시정 조치에 대한 이론적 근거와 설명으로 수집됩니다.
❉ ❉ ❉
PMCF & PSUR
- PMCF(Post-Market Clinical Follow-up)이란, PMS의 한 종류로 임상 평가에 대한 자료를 수집하고 업데이트 하는 프로세스로 MDR의 필수 요구사항입니다. 의료기기의 성능과 안전, 잔류위험에 대한 장기적인 데이터를 수집하고 입증하기 위해 고안되었습니다. 제조업체는 PMCF를 통해 얻은 임상 데이터와 PMS 보고서, 임상 평가 보고서, 리스크 평가 보고서 등의 데이터를 제출하여야 합니다.
- 의료기기의 안전 및 임상 성능을 확인하고, 식별된 위험을 지속적으로 관리하며, 사실적인 증거에 입각한 위험을 감지하는 것이 PMCF의 목적입니다.
PMCF는 새로운 임상 연구, 의료기기 레지스트리에서 파생된 데이터 검토, 시판 전 임상시험을 진행한 고객의 후속조치 또는 이전에 의료기기를 사용했던 환자의 후속조치 등을 통해 이루어질 수 있습니다.
- PSUR(Periodic Safety Update Report)이란, 승인된 의료기기에 대한 안전성 및 성능에 대한 평가를 제공하기 위한 문서입니다. 안정성 및 성능에 대한 새로운 위험 등을 식별하여 포괄적이고 비판적인 분석을 제시하는 것이 PSUR의 목적입니다.
- Class IIb, Class III 등급의 의료기기를 제조하는 업체는 매년 PSUR을 제출해야 하며, Class IIa 등급의 의료기기를 제조하는 업체는 최소 2년에 한번 PSUR을 제출해야 합니다.
- 업체는 PMS 데이터 분석에 대한 보고서를 인증기관에 제출해야 하며, 인증기관은 보고서를 검토한 이후 그 결과를 EUDAMED에 등재하여야 합니다.
❉ ❉ ❉
IGC인증원의 역량
-
IGC는 유럽의 크로아티아에 본사를 두고 있는 DNA Technologies Pacific과의 협력을 바탕으로 임상시험 실시에 대한 서비스를 제공합니다.
-
DNA Technologies Pacific은 임상시험 수탁기관으로서 CRO 업무를 수행하며, 조사에 따른 연구의 결과를 접목시켜 평가에 따른 임상 평가 보고서, PMCF 계획, PMCF 보고서 및 정기 안전 업데이트 보고서 작성을 지원하여 인증 획득 및 인증 유지를 효과적으로 수행할 수 있도록 돕고 있습니다.
❉ ❉ ❉
CE IVD/IVDR | 체외진단용 의료기기 / 유럽 제품인증 소개
체외진단용 의료기기 지침 (In-vitro diagnostic medical devices Directive 98/79/EC, IVD)은 1998년 12월 7일 제정되어 2003년 12월 7일부터 유럽시장에서 판매되는 모든 체외 진단용 의료기기에 적용되었습니다. 이 지침에 적합한 기기에 대해서는 CE 마크가 부착되며, EU는 체외진단 의료기기에 CE 마크 부착을 의무화하고 있습니다.
유럽연합은 2017년 의료기기 ‘규정 (regulation)’을 제정하여 의료기기에 관한 법제를 전면 개정하였습니다. 기존 ‘지침 (directive)’의 형식과 달리 ‘규정 (regulation)’은 직접 회원국에 적용되는 법적 효력을 가지며 회원국의 이행입법을 통하지 않고 직접 적용됩니다.
체외진단 의료기기 규정 (EU Regulation 2017/746, IVDR)은 2017년 5월 26일 발효되었으며, 5년의 경과기간 (transition period)을 거쳐 2022년 5월 26일부터 체외진단 의료기기 규정이 강제 적용됩니다.
체외진단의료기기의 이해
-
체외진단 의료기기 규정은 IVDR Article 2 ‘체외진단 의료기기 정의’에 부합하는 기기에 적용됩니다. 체외진단 의료기기란 단독 또는 조합으로 다음 중 하나 이상에 대한 정보를 제공할 목적으로 인체에서 유래한 혈액 및 조직 기증을 포함한 표본 검사를 위해 체외에서 사용하도록 제조자가 의도한 시약, 시약제품, 측정기, 제어 물질, 키트, 기기, 기구, 장비, 소프트웨어 및 시스템인 의료기기를 의미합니다.
(a) 생리적 또는 병리학적 과정 또는 상태에 관한 것;
(b) 선천적 신체적 또는 정신적 장애에 관한 것;
(c) 의학적 상태 또는 질병의 소인에 관한 것;
(d) 잠재적 대상자와의 안전성 및 호환성을 결정하기 위해;
(e) 치료 반응 또는 작용을 예측하기 위해;
(f) 치료 조치를 정하거나 또는 모니터링하기 위해;
표본 용기도 체외진단 의료기기로 간주됩니다.
-
인증받고자 하는 제품의 의도된 사용 목적이 법령에서 규정하는 체외진단 의료기기 정의에 부합하고 IVDR 요구사항을 충족할 경우 CE Marking을 부착할 수 있으며, 유럽경제지역에 의료기기를 판매할 수 있습니다.
❉ ❉ ❉
IVD/IVDR 주요 변경 사항
-
IVD는 24개 조, 10개 부속서로 구성되어 있으며, IVDR은 10개 장, 113개 조, 15개 부속서로 구성되어 있습니다. IVD가 IVDR로 대체되면서 요구사항이 추가되었으며 제품에 대한 관리가 강화되었습니다.
-
주요 변경 사항은 아래와 같습니다.
| 주요 변경 사항 |
세부 내용 |
| 의료기기 등급 분류 체계 변동 |
IVD에서는 General, Self-testing, List B, List A로 등급을 분류하였지만, IVDR에서는 A, B, C, D로 등급 분류체계가 변경되었습니다. |
| |
| |
IVD 분류 |
IVDR 분류 |
위험도 |
|
| General |
A |
저 |
| Self-testing |
B |
↓ |
| List B |
C |
고 |
| List A |
D |
|
| |
| 적합성 평가 절차의 변동 |
Class A의 경우 인증기관을 통한 적합성 평가를 거치지 않아도 됩니다. 단, Class A 이상의 기기 혹은 Class A 기기 중 멸균되는 기기 (Class As)는 인증기관을 거쳐야 하며 이와 더불어 품질관리 시스템도 요구됩니다. |
| 경제운영자들의 의무사항 변동 |
경제운영자들 (제조업자, 위임대리인, 수입업자, 유통업자)의 의무가 강화 및 추가되었습니다. |
| 성능 평가에 관한 변동 |
IVDR에서는 성능에 대한 정확하고 추가적인 자료 작성이 요구되며 과학적 유효성, 분석 성능, 임상 성능에 대한 내용이 포함되어야 합니다. |
| Class D의 경우, EU 기준 시험소에 의한 시험이 요구됩니다. |
| 고유식별코드 (UDI) 도입 |
식별 및 추적성 확보와 시판 후 안전 확보를 위하여 고유식별코드 (UDI)를 도입하여야 합니다. |
| 소프트웨어에 관한 요구사항 |
IVD에는 포함되지 않았던 소프트웨어 관련 규정이 강화되었습니다. |
❉ ❉ ❉
우리가 제공하는 관련 서비스
IGC는 고객의 해외시장 진출 및 경쟁력 제고를 위한 여러 분야의 인증서비스 및 시험서비스를 제공합니다.
01경영시스템 인증
02제품인증 서비스
03제품 시험
04심사자격 인증 서비스
05심사원 교육 및 양성
CE LVD/EMC | 저전압기기/전자파적합성 유럽 제품인증 소개
CE LVD/EMC란?
유럽의 저 전압 지침 (Low Voltage Directive: LVD)은 특정 전압 범위 내에서 전기 장비의 감전 및 기타 전기 위험에 대한 보호 지침입니다. 이 지침은 75 ~ 1500 V 사이의 직류 전류와 50 ~ 1000 V 사이의 교류 전류의 전압 규격으로 설계, 사용되는 전기 장비를 대상으로 적용됩니다. 또한 해당 지침에 적용되는 전압 규격은 장비 내부의 전압이 아닌 전기 입출력 전압에 관한 것입니다.
저 전압 지침은 전기 장비의 전자기장(Electromagnetic Fields) 방사가 인체에 미치는 영향에 대해서도 정의합니다. 저 전압 지침은 "New" 또는 "Global" 개념이 도입되기 전에 제정된 가장 오래된 단일 지침 중의 하나로 넓은 의미에서 장비에 대한 시장 출시 전 적합성 평가 절차와 장비가 직접 또는 조화 표준 (Harmonized Standards)에 의해 충족해야 하는 필수 보건 및 안전 요구 사항 (Essential Health and Safety Requirements: HSR)을 제공한다고 할 수 있으며 이를 통해 해당 전기 장비가 안전하게 사용될 수 있도록 합니다.
이것은 예측 또는 예측하지 못한 상황에서 발생할 수 있는 다음과 같은 위험요소(Hazard)로부터 인체 및 재산상의 상해나 손실을 방지하기 위한 것입니다.

적용 가능한 위해요소(Hazard)
1) 전기적 위험요소(Electrical Hazard) : 감전 등의 사용중 위험에 노출될 경우
2) 화재적 위험요소(Fire Hazard) : 과부하, 단락 등으로 인한 화재 발생 가능의 경우
3) 기계적 위험요소(Mechnical Hazard) : 기계적 오작동에 의한 위험요소
4) 물리적 위험요소(Physical Hazard) : 낙하, 외부충격, 쓰러짐 등의 구조상의 위험요소
5) 화학적 위험요소(Chemical Hazard) : 제품에 사용한 페인트에 의한 발생 가능 위해요소
CE LVD / EMC 인증절차
⁘ LVD 인증범위
2014/35/EU에 따르면 AC50~1000V 또는 DC75~1500V의 정격전압을 가진 전기제품에 적용합니다. (단, Annex II에 열거된 제품은 제외(방폭기기, 방사선 및 의료기기, 승강기의 전기부품, 전기계량기, 옥내용 플러그 및 소켓 outlet, 전기 철조망 제어기, 전파 방해 기기, 국제기구에서 작성한 안전규정에 부합한 선박,항공기,철도에 사용되는 특수전기기기))
상세 절차
-
[제1단계]
1. 제조자는 CE marking을 진행하기 위해서는 marking하고자 하는 제품을 규정하고 있는 EMC 지침을 파악해야 한다.
EMC 지침: 2014/30/EU Electromagnetic compatibility (EMC) Directive
2. 제품의 적합성을 위해 관련지침(Directive)내에서 규정하고 있는 필수 요건 사항을 항목별로 정리
관련 지침(Directive)에서 규정하는 요구사항의 적합함을 제조자는 증명해야 하며 이를 위해 제품 사용상 안전성 및 위험방지를 위해 대응한 기술적인 내용을 기술하거나 제품시험을 실시한 결과 및 관련 자료를 제시해야 한다.
3. 제조자는 CE Marking 관련 제품 해당지침의 요구사항을 만족하는 사실을 증명, 선언하기 위한 적합성 평가방식(적용 모듈)을 결정하고 그에 따라 적합성 평가를 실시한다.
CE marking 적합성 평가는 8가지 모듈 중 제품의 특성에 따라 하나 또는 두가지 모듈을 결합시킨 형태를 사용하도록 각 지침(Directive)에 규정하고 있다.
4. 제품에 적용할 지침이 결정되었다면 해당지침별로 시험할 유럽 시험규격들을 결정해야 한다.
-
[제2단계]
1. 제품에 관련된 유럽규격(EN Standard)이 결정되었다면 규격에 따라 공인된 기관에서의 시험을 실시한다.
2. 제품시험은 지침에서 요구하는 제품의 안전성 및 위험에 대한 대응 기술을 증명하기 위한 자료로 사용된다.
-
[제3단계]
1. 적합성 선언을 위하여 제품에 대한 안전성 및 위험 방지를 위한 기술적인 내용을 다루는 기술문서(Technical Construction Files; T.C.F)를 작성한다.
2. 기술문서에서 다루어야 할 기술적인 내용들은 해당지침(Directive)에 따라 요구되는 자료가 다르므로 해당지침을 참조하여 준비해야 한다.
-
[제4단계]
이 단계에서는 지침(Directive)의 적합함을 선언하기위해 기술문서 등 여러 기술자료 및 모듈 별로 필요한 품질시스템 인증을 근거로 제조자는 적합성 선언서를 작성하고 그에 대한 확증으로 서명하여 승인한다.
-
[제5단계]
1. 적합성 선언서를 작성하고 승인한 후 제조자는 제품 출하 시 제품의 표시사항을 부착하게 되며 라벨에는 생산자 성명 및 제품 관련 사항과 CE marking을 포함한다.
2. 일반적으로 기술문서를 구성하는 내역은 다음과 같다.
o 제품에 대한 일반사항 및 제품의 작동 체제의 이해에 필요한 내용 및 설명
o 설치도면, 설계도면, 회로도, 회로 계통도 등
o 부품List, 시험성적서, 설계계산, 규격 checklist
o 임상시험 data(MDD 경우), 위험요소 분석
o 중요부품이나 유지보수 시 교환되는 부품 등의 카탈로그, 기술자료 등
-
유럽연합국내에서 인체 및 동물의 안전성을 위협하는 전기 제품이 판매되지 못하도록 하여 전기로부터의 안전성 확보가 이 지침의 목적이며 관련 제품에는 컴퓨터, 정보기술 기기, 가전제품, 동력공구, 시험실 기기, 시험 및 측정 기기와 전원 공급기기가 포함됩니다. 즉, 유럽으로 전기 제품을 판매하고자 하는 제조사에게는 필수적입니다.
CE LVD/EMC 인증의 사후관리
⁘ CE마킹을 한 제품은 일단 EEA 17개국으로 아무런 제약 없이 통관되며 통관된 제품은 유럽 시장에서 아무런 차별 없이 유통될 수 있습니다.
⁘ 세관에서는 CE마킹에 대한 확인과 적합성 선언서를 접수하여 통관시킵니다.
⁘ 단, 통관된 제품인 경우에도 필요한 경우 관련 이해 관계자 (소비자, 경쟁업체 및 자국내 검사기관 등)의 이의신청에 따라 기술문서의 제출요구와 더불어 샘플검사 등 사후관리를 받게 됩니다.
⁘ CE마킹은 강제규격이나 회원국 정부의 CE마킹 주관기관의 사전 검사와 승인을 받아야만 시판할 수 있는 사전 검사제도가 아니라 사후 관리를 받는 제도로써 주관기관은 자발적으로 또는 이해관계자의 신고 또는 문제 발생시 시중 유통제품을 수거하여 규격 적합성 관련 서류검사와 필요시 제품의 안전 검사를 실시하게 됩니다.

우리가 제공하는 관련 서비스
IGC는 다음과 같은 인증 및 관련 서비스를 제공하고 있습니다.
01경영시스템 인증
02제품인증 서비스
03제품 시험
04심사 자격 인증 서비스
05심사원 교육 및 양성
CE Machinery | 기계류 / 유럽 제품인증 소개
유럽 내에서 제품을 유통 또는 판매하고자 하는 제조업체는 그 제품이 해당하는 EU 지침에 적합하다는 것을 적합성 선언 또는 적합성 인증을 통해 CE 마크를 제품에 부착하여야 합니다. 유럽에서 기계류의 안전에 적용되는 지침은 Machinery Directive 2006/42/EC이며, 운반 기계, 기계 장치, 승강기 및 운송장비와 안전 부품을 포함한 모든 기계류에 적용됩니다.
CE Machinery(기계류 안전) 유럽 제품인증 개요
-
기계류의 정의는 “적어도 하나의 부분이 가동부분으로서 구비되어 있으며, 복수의 서로 관계 있는 부품, 부분품이 다 갖추고 적절한 가동기, 제어기 및 동력회로가 있는 것”이라고 MD에 규정되어 있습니다. 유럽의 기계류 지침 (Machinery Directive:2006/42/EC)은 다양한 기계류 및 부분적인 기계류, 부속품, 체인, 로프 및 웨빙, 교체 장비, 이동식 기계 전송장치 및 안전 부품을 포함한 다양한 제품의 사용에 작업자의 건강 및 안전을 보장하기 위해 제정되었습니다.
-
CE 마크를 부착할 수 있는 제품은 다음과 같습니다.
1. 일반 산업용 기계: 반도체 장비, 굴삭기 장착 부속품류 및 부분 기계, 프레스, CNC 선반, 공작기계, 산업용 콤프레셔, 산업용 호이스트, 산업용 프린터, 포장 기기 등
2. 위험 기계류: 원형 톱 등 유사 기계류, 프레스, 플라스틱, 고무 사출기, 광산 등 지하 작업용 제품, 작업용 카 리프트, 승객 운반용 리프트, 안전 릴레이류 등
❉ ❉ ❉
CE Machinery Directive(2006/42/EC)
❀ CE Machinery Directive(2006/42/EEC)의 구성
-
서 론 : 작성에 해당되는 지침과 전반적인 해설 및 유의사항
본 문 : 지침을 적용하기 위한 조문, 가맹국에 대한 법적 의무나 권한
✧ Annex I : 설계 및 제조에 관한 건강과 안전의 필수 요구사항
✧ Annex II : 적합성 선언서에 관한 사항
✧ Annex III : CE – Marking에 관한 사항
✧ Annex IV : Notified Body로부터 EU 형식시험을 받아야 하는 기계류 목록
✧ Annex V : 적합성 선언 및 기술파일
✧ Annex VI : EU 형식시험
✧ Annex VII : Notified Body 기준
❀ 규격 구분
기계 안전에 관한 유럽 규격은 A규격, B 규격, C 규격으로 나눌 수 있습니다. 제품은 해당되는 모든 3종류의 규격을 충족해야 합니다.
❄ 규격 A (EN 414, EN 12100)
기본 안전규격으로 기본 개념, 설계 원칙 및 일반적 측면을 규정하는 규격
❄ 규격 B (EN 13850, EN 13849-1, EN 60204-1 등)
일반적인 기계의 용도 및 목적에 따라 광범위하게 구분된 기계에 적용되는 안전에 관계되는 규격
1) B1규격은 특정 안전 측면에 관한 규격(전기, 안전거리, 표면온도, 소음 등)
2) B2규격은 안전관련 장치 와 기계부품 등에 관한 규격(비상스위치, 압력감지장치 등)
❄ 규격 C
특정 기계 또는 기계 그룹에 관한 상세한 안전 요구사항을 규정하는 규격
❀ 적용 대상 기기
1. 전기, 공기압 및 유압, 무게 등을 에너지원으로 하여 적어도 1개 이상의 작동부분이 있는 제품
2. 건강이나 안전에 관여하는 기계류의 부품으로, 단독으로 출하되는 기기

❀ CE Machinery Directive(2006/42/EC) 핵심 요구사항
제조자가 준수하여야 하는 문서화 요구사항은 MD의 부속서 I의 필수 보건 및 안전 요구 사항에 있으며, 기계가 안전하게 사용되고 있는지, 잔여 위험이 남아있는 경우 사용자에게 올바르게 경고하고 있는지에 대한 내용을 문서화한 것입니다.
필수 요구사항은 다음과 같습니다.
1. 안전에 대한 원칙, 취급을 용이하게 하는 설계, 인체 공학, 제어 시스템, 기계적 위험으로 보호, 전기 및 기타 에너지 온도, 화재 및 폭발 등의 기타 위험, 소음 방출 등 모든 유형의 기계에 공통적인 일반사항, 진동, 방사선 및 유해물질, 유지보수, 청소, 정보의 표시 및 경고
2. 식품, 화장품 및 의약품 등과 같은 특정 등급의 기계류에 적용되는 요구사항
3. 기계의 이동성으로 인한 위험을 상쇄하기 위한 요구사항
4. 리프트 작업의 위험을 상쇄하기 위한 요구사항
5. 지하작업용 기계에 대한 요구사항
6. 기계 리프트 인력에 대한 요구사항
❀ 인증절차
다음과 같이 4가지의 방식이 있습니다.
A) Annex 4에 기술된 고 위험군 기계류
승인기관 (Notified Body : NB)을 통한 인증취득
B) Annex 4가 아닌 일반 기계류 (Annex Ⅱ A)
자가 적합성 선언(완성품에 대한 적합성의 입증으로써 CE 마크를 부착)으로 취득
C) 기계 부품류에 관한 자기 적합성 선언(Annex Ⅱ B)
자가 적합성 선언(완성품에 대한 적합성의 입증으로써 CE 마크를 부착)으로 취득
D) 안전 부품류에 관한 자기 적합성 선언 (Annex Ⅱ C)
자가 적합성 선언(완성품에 대한 적합성의 입증으로써 CE 마크를 부착)으로 취득
참고: B)와 C)의 경우, 자기 적합성 선언서를 작성하고 관련 기술문서를 보관함으로써 규격에 적합함을 선언할 수 있지만 해당제품이나 포장 등에 CE 마크를 부착할 수 없습니다.
❉ ❉ ❉
IGC인증원의 역량
- IGC는 유럽의 인증기관(NB: Notified Body)와 협력을 통해 제품인증을 진행하고 있으며, CE Machinery, CE MDD, CE IVD, CE LVD 등의 제품인증 서비스를 제공하고 있습니다.
- IGC 인증원은 다년간 쌓아온 기술력 및 전문성과 더불어 전세계 주요 시장에서 다양한 특정 범위 및 법적 요구사항에 대한 최신 지식을 보유하고 있습니다.
- 고객 여러분의 전체 글로벌 운영을 지원하기 위한 지식 및 서비스를 제공하고 있습니다.
❉ ❉ ❉
CE PED | 압력장비 / 유럽 제품인증 소개
PED란 Pressure Equipment Directive(2014/68/EU)의 약어로써, 압력장비 지침으로 최대 허용 압력이 0.5bar 이상인 압력장비 및 부품의 설계, 제조 및 적합성 평가에 적용됩니다. EU 국가에 해당 제품의 수출을 원하는 제조자는 압력장비 지침에 따라 인증을 받아야 하며, 압력장비 지침은 2016년 7월 19일자로 97/23/EC에서 2014/68/EU로 개정되었습니다.
인증범위
-
이 지침은 최대 허용 압력이 0.5bar 이상인 압력 장비 및 조립품의 설계, 제조 및 적합성 평가에 적용됩니다. 선박, 배관, 안전 부속품 및 압력 부속품이 모두 포함됩니다.
적용 압력 장비
☑ 수관 보일러
☑ 열교환기
☑ 압력용기
☑ 파이프
☑ 전기 난방기
☑ 밸브(절연, 제어)
☑ 미터
☑ 방출 밸브
☑ 안전 밸브
☑ 제어된 안전 압력 방출 시스템
☑ 압력 스위치
☑ 온도 전환
☑ 유체 레벨 스위치
적용 제외 품목
☑ 단순 압력용기 지침(Simple Pressure Vessels Directive, SPVD)과 이동용 압력용기 지침(Transportable Pressure Equipment Directive, TPED)에 적용되는 품목
☑ 상대적으로 가압이 낮은 제품: 기계지침(Machinery Directive)
☑ 압력용기
☑ 차량용 장비 등
❉ ❉ ❉
압력 기기의 입증 (CE marking)
-
최대 허용 압력(PS), 사용 유체(fluid), 내용적(Volume) 및 외경(DN)에 따라 압력 기기는 Category I, II, III, IV의 4단계로 분류되어, 아래의 표와 같이 해당 category에서 적용 가능한 Module 요구 사항에 따라 인증 절차를 진행합니다. 적합성 평가 시 세부 기술 요구 사항에 대해서는 EN 13445, AD2000 등의 유럽 기술 규격과 ASME code 등을 적용합니다.
-
Annex II, 적합성 평가 테이블
Category |
Modules |
I |
Module A |
II |
Modules A2, D1, E1 |
III |
Modules B(Design type)+D, B(Design type)+F, B(Production type)+E, B(Production type)+C2, H |
IV |
Modules B(Production type)+D, B(Production type)+F, G, H1 |
- Annex III, 적합성 평가 절차
Module A |
내부생산관리 |
Module A2 |
내부생산관리+무작위 주기로 감독 하에 제품 검사 |
Module B |
EU-형식검사(Production type)
EU-형식검사(Design type) |
Module C2 |
내부생산관리에 기반한 형식적합성+무작위 주기로 감독 하에 제품 검사 |
Module D/D1 |
D: 생산공정의 품질보증에 기반한 형식적합성
D1: 생산공정의 품질보증 |
Module E/E1 |
E: 제품 품질보증에 기반한 형식적합성
E1: 최종 제품의 검사 및 시험의 품질보증 |
Module F |
제품검증에 기반한 형식적합성 |
Module G |
단위검증에 기반한 적합성 |
Module H/H1 |
H: 완전품질보증에 기반한 적합성
H1: 완전품질보증에 기반한 적합성+설계검사 |
❉ ❉ ❉
IGC인증원의 역량
♕ IGC는 다년간의 축적된 경험을 바탕으로 유럽에 제품을 수출하고자 하는 기업을 대상으로 다양한 분야에 대한 제품인증 서비스를 제공하고 있습니다.
♕ 해외 유수의 제품인증 기관과 협력관계 구축을 통하여 가장 경쟁력 있는 제품인증 서비스를 제공하고 있습니다.
♕ ISO 9000, 품질경영시스템을 이미 준수하는 기업은 현재의 품질 시스템을 최대한 활용할 수 있고 이를 기반으로 PED의 필수 안전 요구사항을 만족시킬 수 있습니다.
♕ 제조업체가 정식으로 품질 경영 시스템 인증을 받지 않았다면 PED QA 접근 방식에 따라 IGC가 인증 기관 서비스를 제공해 드립니다.
❉ ❉ ❉
우리가 제공하는 관련 서비스
01경영시스템 인증
02제품 인증 서비스
03제품 시험
04심사자격 인증 서비스
05심사원 교육 및 양성
CE SPVD | 단순압력용기 유럽 제품인증 소개
SPVD란 단순압력용기 지침(Simple Pressure Vessels Directive)으로 단순압력용기의 내부압력이 0.5이상인 용접 용기로 공기나 질소를 함유하고 있으며 비화성인 모든 유체를 대상으로 하는 압력용기에 적용되며, 연속적으로 제조되는 단순압력용기에 적용됩니다.
EU 국가에 해당 제품의 수출을 원하는 제조자는 단순압력용기 지침에 따라 인증을 받아야 하며, 단순압력용기 지침은 2016년 4월 20일자로 2014/29/EU로 개정되었습니다.
적용품목/적용 제외 품목
1.적용품목
압력용기의 내부 압력이 0.5 bar 이상인 용접용기로 공기나 질소를 함유하며 비화성인 모든 압력용기
⚜ 압력을 받는 용기의 강도에 영향을 미치는 부위와 조립부품들은 비합금 재질의 강재, 비합금 알루미늄 또는 노화가 일어나지 않는 견고한 알루미늄합금으로 만들어져야 합니다.
⚜ 용기는 바깥쪽으로는 경판으로 닫혀진 원형 단면적의 원통형 부분 또는 원통형 부분과 동일축으로 회전하는 평판 혹은 동일축으로 회전하는 경판으로 구성됩니다.
⚜ 용기의 최대운전압력은 30 bar 를 초과하지 않아야 하며, 용기의 내용적과 압력의 곱은 10,000 bar/liter를 초과하지 않아야 합니다.
⚜ 용기의 최저운전온도는 -50℃보다 낮아서는 안되며 최대운전온도는 강재의 경우 300℃, 알루미늄과 알루미늄합금 용기의 경우는 100℃보다 높지 않아야 합니다.
2. 적용 제외 품목
⚜ 원자력에 사용될 목적으로 방사능 누출이 일어나지 않게 제작된 용기
⚜ 선박이나 항공기의 추진장치 또는 설비에 사용되는 용기
⚜ 소화기
❉ ❉ ❉
필수 요건
SPVD의 필수 요건은 다음과 같습니다:
1) 최대허용압력(PS) x 용량(V)이 50 bar.L를 초과하는 용기는 Annex I의 필수 안전 요구사항을 만족해야 합니다.
2) 최대허용압력(PS) x 용량(V)이 50 bar.L 이하인 용기는 회원국 중 한 국가의 공학적 관행에 따라 설계되고 제조되어야 합니다.
❉ ❉ ❉
SPVD의 중요성
압력용기가 올바르지 않게 취급 및 평가되는 경우, 위험이 초래됩니다. 따라서 유럽지침 2014/29/EU(SPVD)은 모든 압력 용기가 인증기관에 의해 인증되어야 한다고 규정합니다.
이 규격은 다음 사항들에 도움을 줍니다.
⚚ 안전성을 포함한 적합성 평가 절차 충족
⚚ 제품 수출에 필요한 선결조건의 충족
⚚ 유럽 사용 규격 및 지침 요구사항 준수
⚚ 제품의 경쟁력 강화
⚚ 내부 생산관리 강화
❉ ❉ ❉
적합성 평가 절차
단순 압력 용기는 최대허용압력(PS) x 용량(V)에 따라 module이 정해집니다.
| Module B + |
Module C1 |
EU-형식 검사 + 내부 생산관리에 기반한 형식적합성 및 감독 하에 제품 테스트 |
| Module C2 |
EU-형식검사 + 내부 생산관리에 기반한 형식적합성 및 무작위 주기로 감도하에 제품 검사 |
| Module C3 |
EU-형식검사 + 내부 생산관리에 기반한 형식접합성 |
❉ ❉ ❉
IGC인증원의 역량
♔ IGC는 유럽의 인정기구인 FQC(First Quality Certification, NB2674)와 협약으로 단순 압력 용기에 대한 인증 서비스를 제공하고 있습니다.
♔ IGC의 심사원은 다년간 쌓아온 기술력 및 전문성을 통해 경영시스템의 적합성을 정확하게 평가하여 고객의 지속적인 발전에 일조하고 있습니다.
♔ IGC는 제품인증의 신뢰할 수 있는 글로벌 리더로서 ISO SPVD은 물론 PED, Hot-water boiler 등에 대한 인증을 제공하고 있습니다.
♔ IGC는 전세계 주요 시장에서 다양한 특정 범위 및 법적 요구 사항에 대한 최신 지식을 보유하고 있습니다.
♔ IGC는 고객 여러분의 전체 글로벌 운영을 지원하기 위한 지식 및 서비스를 제공하고 있습니다.
우리가 제공하는 관련 서비스
IGC는 SPVD 외 다음과 같은 유럽 제품 인증 서비스를 제공하고 있습니다.
01경영시스템 인증
02제품 인증 서비스
03제품 시험
04심사자격 인증 서비스
05심사원 교육 및 양성
Hot-water boiler / 유럽 제품인증 소개
92/42/EEC는 정격 출력이 4 kW 이상 400 kW 이하인 액체 또는 기체 연료로 연소되는 새로운 온수 보일러의 효율성 요건에 대한 지침으로, 유럽 연합의 에너지 효율 촉진에 관한 SAVE 프로그램의 일부에 의거합니다.
이 지침의 목적은 액체 또는 가스로 연소되는 온수 보일러에 효율 요건을 설정함으로써 에너지 효율을 개선하기 위해 만들어졌습니다.
적용 품목
4 Kw이상 400 Kw 이하의 정격 출력을 가지며 액체 또는 기체 연료를 사용하는 온수 용 보일러 (표준 보일러, 저온 보일러, 가스 콘덴싱 보일러)
❉ ❉ ❉
적용 제외 품목
⚙ 고체연료를 포함한 다양한 연료에 의해 연소될 수 있는 온수 보일러
⚙ 온수 즉석 조제 장비
⚙ 일반적으로 판매되는 액체 및 가스 연료(산업 폐기물 가스, 바이오가스 등)의 특징과는 다른 특성을 가진 연료에 의해 연소되도록 설계된 보일러
⚙ 구내 난방이 주목적이며 중앙난방 및 위생온수를 공급하기 위해 건물에 설치된 조리기구 및 기기
⚙ 중력순환을 사용하여 6kW 미만인 정격 출력을 가지며 보관된 위생온수 생산을 위해 설계된 기기
⚙ 일회용으로 제조된 보일러
❉ ❉ ❉
효율성 요구사항
⛭ 정격 출력: kW로 표시되는 정격 출력(Pn)에서 평균 70 °C의 보일러 수온으로 작동
⛭ 부분 하중: 보일러 유형에 따라 달라지는 보일러의 평균 수온에서 30%의 하중으로 작동
| 보일러 종류 |
출력 범위 |
정격 출력에서의 효율성 |
부분 하중에서의 효율성 |
| |
Kw |
평균 보일러 수온(in ℃) |
효율성 요구사항(in %) |
평균 보일러 수온(in ℃) |
효율성 요구사항(in %) |
| 표준 보일러 |
4 - 400 |
70 |
≥ 84 + 2
logPn |
≥ 50 |
≥ 80 + 3 |
| 저온 보일러 + |
4 - 400 |
70 |
≥ 87.5 + 1.5
logPn |
40 |
≥ 87.5 + 1.5 |
| 가스 콘덴싱 보일러 |
4 - 400 |
70 |
≥91 + 1
logPn |
30(**) |
≥ 97 + 1
logPn |
(*) 액체 연료를 사용하는 콘덴싱 보일러를 포함
(**) 보일러 급수 온도
❉ ❉ ❉
적합성 평가 절차
| Module B + |
Module C |
EU-형식검사 + 유형 적합성 |
| Module D |
EU-형식검사 + 생산 품질 보증 |
| Module E |
EU-형식검사 + 제품 품질 보증 |
❉ ❉ ❉
IGC인증원의 역량
⛯ IGC는 미국의 인정기구인 IAS로부터 인정을 받아 경영시스템에 대한 인증 서비스를 제공하고 있습니다.
⛯ IGC의 심사원은 다년간 쌓아온 기술력 및 전문성을 통해 경영시스템의 적합성을 정확하게 평가하여 고객의 지속적인 발전에 일조하고 있습니다.
⛯ IGC는 경영시스템 인증의 신뢰할 수 있는 글로벌 리더로서 품질, 환경 및 기타 경영시스템에 대한 인증을 제공하고 있습니다.
⛯ IGC는 전세계 주요 시장에서 다양한 특정 범위 및 법적 요구 사항에 대한 최신 지식을 보유하고 있습니다.
⛯ IGC는 고객 여러분의 전체 글로벌 운영을 지원하기 위한 지식 및 서비스를 제공하고 있습니다.
우리가 제공하는 관련 서비스
01경영시스템 인증
02제품인증 서비스
03제품 시험
04심사자격 인증 서비스
05심사원 교육 및 양성
Gas Appliance | 가스 연료 연소기기
유럽 제품인증 소개
Appliances burning gaseous fuels (EU 2016/426) 개요
EU 2016/426은 유럽의 가스기기 규정으로 온수 생산, 냉장, 조명, 난방, 세탁, 조리의 목적으로 제작된 가스용품 및 부속품에 적용됩니다.
유럽의 ‘가스기기 지침(Gas Appliances Directive, GAD 2009/142/EC)’은 2017년 3월 발행된 ‘가스기기 규정(Gas Appliances Regulation, GAR 2016/426)’으로 대체되어 2018년 4월부터 적용되었습니다.
적용 품목
가스를 연료로 하는 조리, 난방, 온수, 냉장, 세탁부분의 기기가 해당되며, 공업용 제품을 제외한 일반적으로 사용되는 가정용 및 상업용 제품
❉ ❉ ❉
적용 제외 품목
⁘ 산업 현장에서 수행되는 산업 공정에서의 사용되는 제품
⁘ 항공기 및 철도에 사용되는 제품
⁘ 실험실에서의 일시적인 사용을 위한 연구 목적의 제품
❉ ❉ ❉
적합성 평가 절차
연속적으로 생산되는 제품의 경우, 제조자는 Module B - Production Type 에따라 Module C2, D, E, F 중 하나를 결합하여 적합성을 평가받아야 합니다.
Module B
(Production Type)
+ |
Module C2 |
EU-형식 검사 + 내부 생산관리에 기반한 형식적합성 및 무작위 주기로 감독하에 제품 검사 |
| Module D |
EU-형식검사 + 생산공정의 품질 보증에 기반한 형식 적합성 |
| Module E |
EU-형식검사 + 제품 품질 보증에 기반한 형식 적합성 |
| Module F |
EU-형식검사 + 제품 검증에 기반한 형식 적합성 |
단일 단위 또는 소량으로 생산되는 제품의 경우에 제조자는 Module G (Unit Verification)로도 적합성을 평가받을 수 있습니다.
❉ ❉ ❉
E-MARK | 자동차인증
유럽 제품인증 소개
E-Mark 인증 개요
EU 자동차 인증은 ECE(Economic Commission for Europe: 유럽경제위원회) 및 EU(European Union: 유럽연합)의 자동차, 관련부품 및 전장품 등을 포함한 자동차의 포괄적인 적합성 규제입니다.
유럽연합 시장으로 자동차 및 자동차 부품을 판매하고자 하는 기업은 관리 감독 기관인 EU 회원국의 교통부로부터 EU 자동차 분야의 승인제도인 E-Mark 인증을 받아야 합니다.
E-Mark 인증을 받은 자동차 또는 자동차 부품만이 유럽연합 내에서 자유롭게 수출 및 거래될 수 있습니다.
적용 품목
일반적으로 자동차, 트럭, 버스, 트레일러 및 기타 도로 주행 차량 (차량 탑재 ESA/부품 포함)은 이제 전자기 호환성에 대한 UNECE 규정 10을 준수해야 합니다. 이에 따라 형식 승인 인증을 받은 시스템 EMC에 대한 UNECE Regulation 10은 대문자 “E” E-Mark로 표시되어 있습니다.
e-Mark 적용 품목
EU지침에 준수하는 차량 및 부품: 스티어링 장치, 브레이크, 각종 램프, 난방 시스템, 속도계, 헬멧, 아동용 안전 의자, 배기관, 도난 방지 장치, 타이어, 안전 벨트, 연결 장치, 자동차용 유리, 냉방 시스템, 저류 보호, 버스의 내장재, 경음기, 시트 및 해드레스트 등.
❉ ❉ ❉
적용 규격
UN-ECE Regulation 10 that replaced the ‘Automotive Electromagnetic Compatibility Directive’ 2004/104/EC (AEMCD) in November 2014. / 2014년 11월 '자동차 전자기 호환성 지침 2004/104/EC (AEMCD)를 대체 한 UN-ECE 규정 10
e-Mark 관련규격
2007/46/EC: approval of motor vehicles and their trailers, and of systems, components and separate technical units intended for such vehicles / 자동차 및 트레일러, 그리고 그러한 차량을 위한 시스템, 부품 및 별도의 기술 장치의 승인
❉ ❉ ❉
E-Mark 와 e-Mark 차이점
E-Mark
E-Mark는 EU에 판매되는 승인된 차량 및 차량 부품에 대한 UN마크로, 자동차의 부품이나 시스템의 형식승인에 대한 인증입니다.
E-Mark는 세계 기준인 ECE Regulation에 적합하다는 것을 의미합니다.
e-Mark
e-Mark는 EU에 판매되는 승인된 차량 및 차량 부품에 대한 EU마크로, 자동차의 부품 및 시스템은 물론 전 차량에 대한 형식승인을 포함하는 인증입니다.
e-Mark는 유럽의 강제 지침인 유럽 자동차 EMC 지침에 적합하다는 것을 의미합니다.
❉ ❉ ❉
인증마크
유럽 경제위원회 (ECE, UN 기관) 인증, E-Mark
 E-Mark 인증마크
E-Mark 인증마크
❖ E-Mark는 유럽 경제위원회에서 발행한 규정입니다.
❖ E-Mark 로고는 원형 프레임입니다.
❖ 번호는 승인을 부여한 EU 국가 또는 주 번호입니다. 한 국가 / 주가 승인하면 다른 모든 회원국에 대해 충분한 것으로 간주됩니다.
❖ 국가 / 주 코드는 다음과 같습니다.
| E1—Germany |
E16—Norway |
E32—Latvia |
E46—Ukraine |
| E2—France |
E17—Finland |
E33— |
E47—South Africa |
| E3—Italy |
E18—Denmark |
E34—Bulgaria |
E48—New Zealand |
| E4—Netherlands |
E19—Romania |
E35— |
E49—Cyprus |
| E5—Sweden |
E20—Poland |
E36—Lithuania |
E50—Malta |
| E6—Belgium |
E21—Portugal |
E37—Turkey |
E51—Korea |
| E7—Hungary |
E22—Russia |
E38— |
E52—Malaysia |
| E8—Czech Republic |
E23—Greece |
E39—Azerbaijan |
E53—Thailand |
| E9—Spain |
E25—Croatia |
E40—Macedonia |
E54— |
| E10—Yugoslavia |
E26—Slovenia |
E41— |
E55— |
| E11—United Kingdom |
E27—Slovakia |
E42—European Community |
E56—Montenegro |
| E12—Austria |
E28—Belarus |
E43—Japan |
E56—Tunisia |
| E13—Luxembourg |
E29—Estonia |
E44— |
|
| E14—Switzerland |
E31—Bosnia |
E45—Australia |
|
EU 인증 (EC), e-Mark
 e-Mark 인증마크
e-Mark 인증마크
❖ e-Mark는 EU 지침을 기반으로한 안전 인증 마크입니다.
❖ EU 집행위원회는 회원국이 자동차, 부품 및 시스템에 적용하도록 요구합니다.
❖ e-Mark 로고는 직사각형 프레임입니다.
❖ 번호는 승인을 부여한 EU국가 또는 주 번호입니다. 한 국가 / 주가 승인하면 다른 모든 회원국에 대해 충분한 것으로 간주됩니다.
❖ 국가 / 주 코드는 다음과 같습니다.
| e1 – Germany |
e11 – United Kingdom |
e24 – Ireland |
| e2 – France |
e12 – Austria |
e26 – Slovenia |
| e3 – Italy |
e13 – Luxembourg |
e27 – Slovakia |
| e4 – Netherlands |
e17 – Finland |
e29 – Estonia |
| e5 – Sweden |
e18 – Denmark |
e32 – Latvia |
| e6 – Belgium |
e19 – Romania |
e34 – Bulgaria |
| e7 – Hungary |
e20 – Poland |
e36 – Lithuania |
| e8 – Czech Republic |
e21 – Portugal |
e49 – South Cyprus |
| e9 – Spain |
e23 – Greece |
e50 – Malta |
❉ ❉ ❉
시험 규격
Regulation 10 Concerning the approval of vehicles with regard to electromagnetic compatibility
규정 10 전자기 호환성과 관련된 차량 승인 관련
| 시험 장비 |
시험항목 |
시험규격 |
| EMI Measurement System |
Radiated Emission |
Regulation No.10, CISPR 25 |
| Conducted Emission (Current) |
| Conducted Emission (Voltage) |
| EMS Test System |
RI Test (ALSE) |
Regulation No.10,ISO 11452-2 |
| BCI Test |
Regulation No.10,ISO 11452-4 |
| Transient |
Transient Emission |
Regulation No.10, ISO 7637-2 |
| Transient Immunity |
Regulation No.10, ISO 7637-2 |
❉ ❉ ❉
인증 절차
1. 신청
신청서 작성 및 제출
2. 문서&샘플
기술문서 및 샘플 준비
3. 시험 실시
샘플 시험 실시
4. 생산 적합성 검사(Conformity of production)
생산공장의 기술기준에 따라 해당제품을 생산할 능력이 있는지를 평가하는 현장검사 실시.
*ISO9000 및 EU 국가 인증 기관에서 발급한 기타 품질 시스템 인증서를 획득 한 제조업체는 공장 검사가 면제될 수도 있습니다.
5. 시험성적서
시험성적서 발행, 기타 기술문서와 함께 유럽 국가 교통부에 제출
6. 인증서 발행
서류심사 및 해당 지침에 대한 시험결과와 제조공장에 대한 현장 감사 결과를 종합적으로 검토하여 합격시 유럽 국가 교통부에서 인증서 발행
7. 정기적 공장심사
발급 기관은 인증된 제품 제조업체의 공장 검사를 정기적 또는 비정기적으로 추적, 검사할 수 있습니다.
*ISO9001을 획득한 공장의 경우 공장검사기간이 단축되거나 경우에 따라서는 면제될 수도 있습니다.
❉ ❉ ❉
준비 사항
제출 문서는 영문으로 작성 및 제출되어야 합니다.
1. 제조사 관련 정보
-제조업체명
-제조업체 주소
-사업자등록증 사본
2. 제품 관련 정보
-제품매뉴얼/사용설명서
-제품도면
-부품 리스트
-부품 배치도
-전기블록 다이어그램
-제품사진
-회로도
3. 제조공장의 ISO 9001 또는 IATF 16949 인증서
❉ ❉ ❉
IGC인증원의 역량
⁙ IGC는 전세계 주요 시장에서 다양한 특정 범위 및 법적 요구 사항에 대한 최신 지식을 보유하고 있으며, 고객 여러분의 전체 글로벌 운영을 지원하기 위한 지식 및 서비스를 제공하고 있습니다.
⁙ IGC는 자동차 분야에서의 고객들에게 전문적이고 경쟁력 있는 서비스를 제공합니다.
⁙ IGC는 유럽의 크로아티아 교통관련 기관과 협력하여 E-Mark 인증 서비스를 제공합니다.
❉ ❉ ❉
