CE MDD | Medical Device
European Product Certification Introduction
The medical Device Directive is intended to harmonize the legislation on medical devices within the European Union. Legally, in order to sell Medical devices to the European Market, manufacturers must comply with the requirements of the Medical Device Directive. The applicant’s product and quality system must be evaluated, and the manufacturer must affix the CE mark before selling the products.
Applicable Items
-
The Medical Device Directive is applicable to devices according to the definition of ‘medical device’. A medical device is a machine, device, equipment, material, or other article used alone or in combination that contains software intended for human use as intended by the manufacturer.
-
Is applicable to diagnosis, prevention, monitoring, treatment or alleviation, diagnosis, monitoring, treatment of disease or alleviation or compensation for injury or disability, investigation, replacement or modification of anatomical or physiological processes, control or concepts. It also refers to something that does not achieve its intended primary action in or on the human body by pharmacological, immunological or metabolic means, but can help its function by such means.
❉ ❉ ❉
Qualification Assessment Process
❀ classification
- Annex IX of the Medical Device Directive(93/42/EEC) provides 18 rules for classifying medical devices. Under these rules, medical devices are classified according to their intended purpose.
✧ Rule 1 ~ 4 : Non-invasive device
✧ Rule 5 ~ 8 : Invasive device
✧ Rule 9 ~ 12 : Active device
✧ Rule 13 ~ 18 : Special Law
- Medical device risk assessment to patients and users are identified and classified according to the above 18 rules. The eligibility assessment process is based on the identified risk values. If the risk value is higher, the eligibility requirements get more stringent.
- Class I medical devices follow the evaluation procedures in Annex VII.
❀ Procedure of Class I Including measurement function)
Manufacturer’s option :
- Qualification and technological documentation of competence to meet metrological requirements for each or statistically selected ample in accordance with Annex VII section 3 and Annex IV
- Evaluation and technical documentation of product quality system in accordance with Annex VII section 3 and Annex V which is related to statistical requirements.
- Quality assessment and technical documentation of medical devices in accordance with Annex VII section 3 and Annex VI associated with statistical requirements.
❀ Procedure of Class I (including sterilization)
Manufacturer’s option :
- Evaluation and technical documentation of product quality systems in accordance with Annex VII section 3 and annex V associated with sterile conditions.
❀ Procedure of Class IIa Medical device
Manufacturer’s option :
- Qualification and technical documentation of each or statistically selected sample according to Annex VII Section 3 and Annex IV Section 8.
- Evaluation and technical documentation of the product quality system in accordance with Annex VII section 3 and Annex V section 6(examined at the manufacturer’s site).
- Evaluation and technical documentation of medical device quality in accordance with Annex VII Section 3 and Annex VI Section 6(examined at on-site audit).
- Evaluation and technical documentation where the overall quality system has been audited at the manufacturer’s site in accordance with Annex VII section3 and Annex II (Development evaluation excluded according to section 4)
❀ Procedure of Class IIb Medical Device
Manufacturer’s option :
- Qualification, type examination and technical documentation for each manufactured product in accordance to Annex III Section 3: Samples selected statistically(randomly) according to Annex IV or validated by Section 5.
- Evaluation, type examination and technical documentation of manufacturing quality systemin accordance with Annex III Section 3 and Annex V(examined at onsite audit)
- Technical documentation of evaluation and type examination of medical device in accordance with Annex III Section 3 and Annex VI(examined at manufacturer’s onsite audit)
- Technical documentation of the evaluation of all quality systems that have been audited on site of the manufacturer in accordance with Annex II Section 3.2 and Annex II (excluding development evaluation by Section 4)
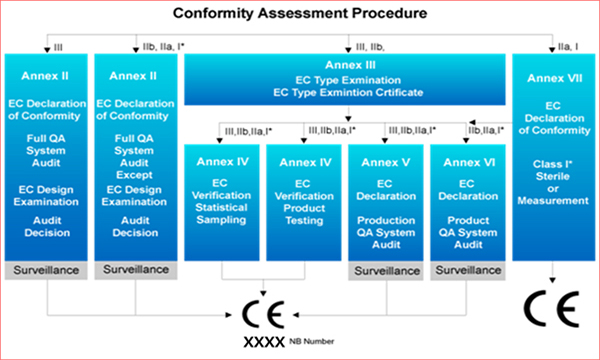 Conformity assessment procedure
Conformity assessment procedure
❀ Procedure of Class III Medical device
Manufacturer’s option :
- Evaluation of all quality systems according to Annex II Section 3.2 and Annex II(examined at the manufacturer’s on site audit) and technical documentation of the description of the development according to Section 4.2.
- Annex III Section 3 and Qualification, Type examination and technical documentation for each manufactured device: Verification shall be made by Section 5 for statistically (randomly) selected samples in accordance with Annex IV Section 6.
- Technical documentation of Evaluation, type examination of the product quality system(examined at the manufacturer’s onsite audit) in accordance with Annex III Section 3 and Annex V Section 3.2
❉ ❉ ❉
End of the MDD
- The European Medical Device Directive MDD(93/42/EEC) will be completely replaced by MDR from 26 May 2020. After that, it is not possible to apply for CE certification to MDD, and the new medical device must meet the MDR (2017/745/EU) requirements.
- However, if the MDD conformity assessment has been completed and the certificate has been issued before the time of compulsory application, the product can be shipped(exported) to European jurisdiction with the validity of the certificate recognized up to May 27, 2024.
- You may check the above materials officially issued by the European Authority.
❉ ❉ ❉
CE / MDD Service
- IGC is currently working on a CE/MDD project in cooperation with 3 Notified Body (referred to as NB) organizations.
- Because of this, you can apply without exception of almost all scopes from Class I to Class III.
- We will not spare any support to obtain your CE/MDD certification based on the experience accumulated through numerous projects.
❉ ❉ ❉
Related Services from IGC
01System certification (ISO 13485, ISO 15378, ISO 14155)
02Product certification (European CE certification, clinical evaluation, medical device registration [Eurasia, China, USA, Thailand, Taiwan])
03Certification of Auditor Qualifications
04Professional manpower training and education
CE MDR | Medical Device
European product certification introduction
What is CE Marking ?
The medical Device Regulation is intended to harmonize the legislation on medical devices within the European Union. Legally, in order to sell Medical devices to the European Market, manufacturers must comply with the requirements of the Medical Device Regulation(MDR) after enforcement in 2020. The applicant’s product and quality system must be evaluated, and the manufacturer must affix the CE mark before selling the products.
MDR overview
- Applicable standards : Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 Ap
- Legal content : Council Directive 93/42/EEC (MDD) + Council Directive 90/385/EEC (AIMD)
- Effective Date : May 25, 2017
- Compulsory application date : May 26, 2020, after the MDR takes effect a grace period of 3 years
- Purpose : To create a safer, more effective and innovative medical device market in Europe.
- MDD certificate final validity date: May 26, 2024 (sale until May 27, 2025)
- Changed from Directive to Regulation. Regulation change more frequently than Directives and are more enforceable.
Major Revision Contents
- Strong control of health authorities over the Notified Body(NB): Reinforcement of sub-contractor management
- Introduction and application Common Specification for each product in addition to the harmonized standard.
- Strengthening the responsibility of economic operators [Manufacturer, Importer, Distributor, Authorized Representative]
- Strengthening the responsibility of economic operators [Manufacturer, Importer, Distributor, Authorized Representative)
- NB’s strong control over manufacturers
- Expansion of clinical evaluation of Pre/Post Market and management of all clinical investigations, and reinforcement of market surveillance (Vigilance & PMS) after marketing by manufacturers
- Establishment of computer system based on EUDAMED(European database on medical devices): product registration, UDI, economic operator registration, NB and certificate, clinical research, Vigilance & PMS, market research (health authorities)
- Improvement and introduction of UDI System : sequential introduction from Class III. Currently it’s compatible with FDD, subject to change.
- MDCG(Medical Device Coordination Group) : A medical device coordination group composed of independent expert within the health authority. Evaluate NB, Manage Class III, Consult clinical evaluation and investigate conformity assessment in detail for Class III implantable& Class IIb active device product.
- Class I reusable surgical instrument : Like Class I sterilization/measuring devices, subject to NB review → only for reprocessing processes such as washing/cleaning process
- Implant Card : Doctor, manufacturer, patient, serial number, etc.
- Person Responsible for Regulatory Compliance) : A person who has more than one year of quality & RA work experience, and four-year college degree or higher, responsible for the release of technical documents and products.
- Application of MDR to products that do not belong to the definition of medical devices → Fillers, ties, and laser surgery devices(hair removal, cosmetic purposes)
❉ ❉ ❉
Content to be included in the TCF(Technical Document)
- Completion of GSPR(General Safety and Performance Requirements) Check List of MDR Annex 1
- Biological Safety Assessment Report (ISO10993-1:2018)
- Risk Management Plan (ISO14971:2019)
- Risk Management Report
- Usability Validation Plan (IEC62366-1:2015)
- Usability Validation Report
- Clinical Evaluation Plan (MEDDEV 2.7/1 :2016
- Clinical Literature Search Protocol
- Clinical Literature Search Report
- Adverse Event Search Report
- Clinical Evaluation Report
- Prepare Process (Sterilization, Cleaning, Packaging, Clean Room) Validation Report
- Software Validation Plan
- Software Validation Report
- Product drawing and schematic drawing
- Manufacturing flow chart
- User Manual (Instructions for use)
- List of used materials(important components), etc.
- Product Label
- Annual upload of SSCP(Summary of safety and clinical performance) : Implantable and Class III Article 32
- Regularly update PSUR(Periodic Safety Update Report) : Class IIa, IIb, III product Article 86
- DOC (Declaration of Conformity)
❉ ❉ ❉
IGC’s Competency
- IGC is contributing to the continuous development of customers by accurately evaluating the suitability of product certifications through the technology and expertise accumulated over the years.
- IGC has up-to-date knowledge of a wide range of specific scopes and legal requirements in major markets around the world and provides the knowledge and services to support your entire global operation.
- In addition, IGC provides various certification related services in the medical device field.
✦ 93/42/EEC
✦ ISO 13485
❉ ❉ ❉
Related Services from IGC
01System certification (ISO 13485, ISO 15378, ISO 14155)
02Product certification (European CE certification, clinical evaluation, medical device registration [Eurasia, China, USA, Thailand, Taiwan])
03Certification of Auditor Qualifications
04Professional manpower training and education
Clinical Evaluation
European Product Certification Introduction
Clinical evaluation is the process of collecting and evaluating clinical data related to medical devices to verify safety, effectiveness and performance, and to demonstrate conformity to relevant regulatory requirements.
Clinical evaluation is a process conducted throughout the life cycle of a medical device, it should be conducted in the process of developing a medical device to approve data for manufacturers to study and obtaining new information about the device’s safety, effectiveness and performance.
Medical device manufacturers shall meet the requirements of ISO 14155 standard when conducting clinical trials in order to prove the safety and effectiveness of the product according to the purpose of use.
MDR and Clinical
- Medical Device Regulation(MDR) refers to a new medical device regulation that all medical device manufacturers planning to enter the European market must comply with. MDR requires a process that enhances clinical data and evaluation processes, and manufacturers must provide clinical evaluation reports to demonstrate the safety and performance of medical devices.
- For Class III and Class IIb products, clinical evaluation is mandatory, and the clinical evaluation instruction manual and PMCF plan are included.
- Clinical data can be acquired by collecting data on the clinical experience of equivalent medical devices, deriving clinical results through clinical evaluation reports, and clinical evaluation using similar devices.
❉ ❉ ❉
Stages of Clinical Evaluation
1. Establish a clinical evaluation plan
2. Creation and identification of clinical data (Literature search, clinical experience, clinical research)
3. Evaluation of the suitability of clinical data
4. Analyzing the entire data and drawing conclusion
5. Preparation of clinical evaluation report
❉ ❉ ❉
Prerequisite of Clinical Trial Data
-
Clinical trial evaluation data should include.
✤ Risk assessment, justification of the designed clinical trial, clinical trial stage, clinical trial brochure, clinical trial
-
Clinical progress report, monitoring plan, reasons for selection of clinical trial region, clinical participant consent, and clinical participant identification method, etc. should be included.
✤ Information about the initial clinical trial region is required.
✤ Periodic monitoring for clinical trial region is required.
✤ Data on side effects or device defects is required.
❉ ❉ ❉
What is PMS?
- ‘PMS’ is also referred to as a post-market surveillance system and an any activity carried out by manufacturers including pre-gather and review experience gained from medical devices placed on the EU market, and to keep the collected data up to date.
- Manufacturers must plan, establish, document, implement, maintain and update post-market surveillance systems for each device in a manner proportional to the risk class and appropriate for the device type. Post-market surveillance systems must be suitable for actively and systemically collecting, recording and analyzing data on the quality, performance and stability of devices over their entire life span, drawing necessary conclusions, and determining, implementing and monitoring preventive and corrective actions.
- In MDR, there is an increased obligation to proactively collect post market requirements and data for manufacturers and to document and report the analysis of these post market activities and data..
❉ ❉ ❉
PMS Report Creation
- Post-Market Surveillance Report (PMSR)
Manufacturers of Class I devices are required to prepare a post-market surveillance report summarizing and analysis results and conclusions of the post-market surveillance data collected as a result of the post-market surveillance plan to prepare a rationale and explanation for prevention and correction. If necessary, the report must be updated and provided to the competent authority upon request.
- Periodic Safety Update Report (PSUR)
Manufacturers of Class IIa, Class IIb and Class III devices are required to summarize the results and conclusions of the post-analysis for each category or group of devices, as well as results of periodic safety update report (PSUR) for each device. Market surveillance data is collected as a rationale and explanation for preventive and corrective actions taken as a result of post market surveillance plans.
❉ ❉ ❉
PMCF & PSUR
- PMCF(Post-Market Clinical Follow-up), a type of PMS, is the process of collecting and updating data on clinical evaluation, which is an essential requirement of MDR. It is designed to collect and validate long-term data on the performance, safety and residual risk of medical devices. Manufacturers are required to submit clinical data obtained through PMCF and data such as PMS reports, clinical assessment reports, and risk assessment reports.
- The purpose of PMCF is to verify the safety and clinical performance of medical devices, to continuously manage identified risks, and to detect risks based on factual evidence. PMCF can be achieved through new clinical studies, review of data derived from medical device registries, follow-up by customers who have conducted premarket clinical trials, or through follow-up by patients who have previously used medical devices.
- The PSUR(Periodic Safety Update Report) is a document intended to provide an evaluation of the safety and performance of approved medical devices. It is the purpose of PSUR to present a comprehensive and critical analysis by identifying new risks to stability and performance.
- Manufacturers of Class IIb, Class III medical devices must submit a PSUR annually, and manufacturers of Class IIa medical devices must submit a PSUR at least once every two years.
- The company must submit a report on PMS data analysis to the certification body, and after reviewing the report, the certification body must register the result in EUDAMED.
❉ ❉ ❉
IGC’s Competency
-
IGC provides services for conducting clinical trials in partnership with DNA Technologies Pacific, headquartered in Croatia, Europe.
-
DNA Technologies Pacific performs CRO as a clinical trial consignee, and supports the creation of clinical evaluation reports, and regular safety update reports according to evaluation by incorporating the results of research from investigation which will help you effectively acquire and maintain certification.
❉ ❉ ❉
Related Services from IGC
01System certification (ISO 13485, ISO 15378, ISO 14155)
02Product certification (European CE certification, clinical evaluation, medical device registration [Eurasia, China, USA, Thailand, Taiwan])
03Certification of Auditor Qualifications
04Professional manpower training and education
CE IVD/IVDR | In-vitro diagnostic medical device
European product certification introduction
The In-vitro diagnostic medical devices Directive 98/79/EC, IVD was enacted on December 7, 1998 and has been applied to all in vitro diagnostic medical devices sold in the European market from December 7, 2003. Devices that comply with this directive are marked with the CE mark, and the EU mandates to attach the CE mark to in vitro diagnostic medical devices.
The European Union enacted the ‘regulation for medical devices in 2017 and revised the legal system on medical devices. Unlike the existing form of ‘Directive’, ‘Regulation’ has legal effect applied directly to member states and is applied directly, not through implementing legislation of member states.
In vitro diagnostic medical device regulation(EU Regulation 2017/746, IVDR) entered into force on May 26, 2018, and the invitro diagnostic medical device regulation was enforced from May 26, 2022 after a transition period of 5 years.
Understanding of in-vitro diagnostic medical devices
In vitro diagnostic medical device regulations are applied to devices that comply with IVDR Article 2 ‘Definition of in vitro diagnostic medical devices’. In vitro diagnostic medical devices, either alone or in combination, mean reagents, reagent products, measuring instruments, kits, instruments, equipment, software and system medical devices intended by the manufacturer to be used in vitro for specimen testing, including donation of blood and tissues derived from human body for the purpose of providing information on one or more of the following:
(a) Relating to physiological or pathological process or condition;
(b) Relating to a congenital physical or mental disorder;
(c) Relating to a medical condition or predisposition to disease;
(d) To determine safety and compatibility with potential subjects;
(e) To predict treatment response or action;
(f) To establish or monitor treatment measures;
Sample containers are also considered in-vitro diagnostic medical devices.
-
If the intended use of the product to be certified meets the definition of in vitro diagnostic medical devices stipulated by laws and meets the IVDR requirements, CE marking can be affixed, and medical devices can be sold in the European Economic Area.
❉ ❉ ❉
Major changes in IVD/IVDR
-
IVD consists of 24 groups and 10 annexes, and IVDR consists of 10 chapters, 113 groups and 15 annexes. As IVD was replaced by IVDR, requirements were added and the management of the product was strengthened.
-
The major changes are as followings:
| Major changes |
Details |
| Medical device classificaiton system change |
In IVD, the grades were classified into General, Self-testing, List B, List A, but in IVDR, the grade classification system was changed to A, B, C, D. |
| |
| |
IVD classification |
IVDR classification |
Risk |
|
| General |
A |
Low |
| Self-testing |
B |
↓ |
| List B |
C |
High |
| List A |
D |
|
| |
| Changes in conformity assessment procedure |
In the case of Class A, there is no need to undergo conformity assessment through a Certification Body. However, devices that are sterilized among Class A(Class As) or higher must go through a certification body, and a quality management system is also required. |
| Changes in the obligation of economic operators |
The obligation of operators(manufacturer, delegated agent, importer, distributor) have been strengthened and added. |
| Changes in performance evaluation |
IVDR requires the preparation of accurate and additional data on performance and should include scientific validity, analytical performance, and clinical performance. |
| For Class D, testing by an EU standard laboratory is required. |
| Introduction of UDI |
A unique identification code(UDI) must be introduced to secure identification and traceability and to secure post-marketing safety. |
| Requirements for the software |
Regulations related to software that were not included in IVD have been strengthened. |
❉ ❉ ❉
Related Services from IGC
01System certification (ISO 13485, ISO 15378, ISO 14155)
02Product certification (European CE certification, clinical evaluation, medical device registration [Eurasia, China, USA, Thailand, Taiwan])
03Certification of Auditor Qualifications
04Professional manpower training and education
CE LVD/EMC | Low Voltage/Electromagnetic Compatibility
European product certification introduction
What is CE LVD/EMC?
The European Low Voltage Directive(LVD) is a directive to protect electrical equipment against electric shock and other electrical hazards within a specific voltage range. These guidelines apply to electrical equipment designed and used with voltage specification for direct current between 70 ~ 1500 V and alternating current between 50 ~ 1000 V. Also, the voltage specifications that apply to these directives are for the electrical input/output voltage, not the voltage inside the equipment.
The Low Voltage Directive also defines the human body’s effects of electromagnetic field radiation from electrical equipment. The Low Voltage Directive is one of the oldest single directives established prior to the introduction of the “New” or “Global” concepts. In a broad sense, it provides a pre-market conformity assessment procedure for a device and the Essential Health and Safety Requirements(HSR) that the device must meet either directly or by harmonized standards, which ensure that equipment is used safely.
This is to prevent personal and property injury or loss from the following hazards that may occur under predicated or unforeseen circumstances.

Applicable Hazard
1) Electrical Hazard : When exposed to danger during use such as electronic shock
2) Fire Hazard : In case of fire due to overload, short circuit, etc.
3) Mechanical Hazard : Risk factors due to mechanical malfunction
4) Physical Hazard : Structural risk factors such as falling, external shock, and collapse
5) Chemical Hazard : Possible hazards caused by the paint used on the product
CE LVD / EMC Certification Procedure
⁘ LVD Certification scope
According to 2014/35/EU, it is applied to electrical appliances with a rated voltage of AC50~1000V or DC75~1500V. (However, products listed in Annex II are excluded(Explosion-proof equipment, radiation and medical equipment, electric parts of elevators, electric meters, indoor plugs and socket outlets, electric barbed wire controllers, jamming equipment, special electricity used for ships, aircraft, and railroads complying with safety regulations prepared by international organization device))
Detailed Procedure
[Step 1]
-
1. In order to carry out CE marking, the manufacturer must understand the EMC Directive that defines the product to be marked.
EMC Directive: 2014/30/EU Electromagnetic compatibility (EMC) Directive
2. For the suitability of the product, the essential requirements stipulated in the Directive shall be organized by item
The manufacturer must prove that the requirements stipulated in the relevant Directive are met. For this purpose, technical details of the response for safety and risk prevention in product must be described or results of product test and related data must be presented.
3. The manufacturer determines the conformity assessment method(applied module) to prove and declare that the product meets the requirements of the relevant guidelines related to CE marking, and conducts conformity assessment accordingly. CE marking conformity assessment stipulates in each Directive to use one or a combination of two modules according to the characteristics of the product among the 8 modules.
4. Once the guidelines to be applied to the product have been determined, the European test standards to be tested for each of those guidelines should be determined.
[Step 2]
-
1. If the EN standard related to the product has been determined, conduct the test in an accredited institution according to the standard.
2. Product tests are used as data to prove the safety and risk response skills of products required by the guidelines.
[Step 3]
-
1. For the declaration of conformity, a technical document (Technical Construction Files; T.C.F) that deals with the technical contents for the safety and risk prevention of the product shall be prepared.
2. The technical contents to be dealt with in the technical document must be prepared by referring to the relevant guidelines as the required data differs according to the relevant directive.
[Step 4]
-
In this step, in order to declare the conformity of the Directive, the manufacturer prepares a declaration of conformity based on the quality system certification required for each module and various technical documents, and signs and approves it as a confirmation.
[Step 5]
-
1. After completing and approving the declaration of conformity, the manufacturer attaches the labeling information of the product when the product is shipped. The label includes the manufacturer’s name, product related matters and CE marking.
2. In general, the details of the technical document are as follows:
o General information about the product and explanation necessary for understanding the operating system of the product
o Installation drawings, design drawings, circuit diagrams, circuit distribution diagram, etc.
o Parts List, test report, design calculation, standard checklist
o Clinical trial data(for MDD), risk factor analysis
o Catalogs, technical data, etc. of important parts or parts exchanged during maintenance
-
The purpose of this guideline is to ensure safety from electricity by preventing the sale of electrical products that threaten human and animal safety in the European Union. Related products include computers, information technology equipment, household appliances, power tools, laboratory equipment, test and measurement equipment and power supplies. In other words, it is essential for manufacturers planning to sell electrical products to Europe.
Post management of CE LVD/EMC certification
⁘ CE marked products are cleared through 17 EEA countries’ custom without any restriction. Cleared products can be distributed in the European market without any discrimination.
⁘ CE Customs checks the CE marking and accepts the declaration of conformity.
⁘ Even for the customs clearance product, if necessary, follow-up management such as sample inspection, along with a request for submission of technical documents, is subject to objections from relevant stakeholders (customers, competitors, and domestic inspection agencies).
⁘ CE marking is a system that receives post-management, not a pre-inspection system that can be marketed after receiving pre-inspection and approval of CE marking authority of the member country’s government or mandatory standard. The CE marking authority will voluntarily, or when a stakeholder reports or a problem occurs, collects the products distributed on the market and conducts a document inspection related to conformity of standards and, if necessary, conducts a safety inspection of the product.

Related Services from IGC
01System certification (ISO 13485, ISO 15378, ISO 14155)
02Product certification (European CE certification, clinical evaluation, medical device registration [Eurasia, China, USA, Thailand, Taiwan])
03Certification of Auditor Qualifications
04Professional manpower training and education
CE Machinery | Machinery
Introduction to European Product Certification
A manufacturer that intends to distribute or sell a product within Europe shall affix a CE mark to the product through declaration or certification of conformity that the product conforms to the applicable EU guidelines. In Europe, the guidelines applicable to the safety of machinery are Machinery Directive 2006/42/EC and apply to all machinery, including transport machines, mechanism, lifts and transport equipment and safety components.
CE Machinery(Machinery Safety) European Product Certification Overview
The definition of machinery is defined in MD as "at least one part of the operation, with multiple interrelated parts and proper actuator, controller and power circuitry." The European Machinery Directive (Machinery Directive:2006/42/EC) is designed to ensure the health and safety of workers in the use of a wide range of machinery and other products, including accessories, chains, ropes and webbing, replacement equipment, mobile mechanical transmitters and safety components.
The following products can be attach CE marks:
1. General industrial machinery: semiconductor equipment, excavator accessories and partial machinery, press, CNC lathe, machine tool, industrial compressor, industrial hoist, industrial printer, packaging equipment, etc.
2. Hazardous machinery: similar machinery, such as circular saws, products for underground work, such as presses, plastics, rubber ejectors, mines, car lifts for work, lifts for passenger transport, safety relays, etc.
❉ ❉ ❉
CE Machinery Directive(2006/42/EC)
❀ Composition of CE Machinery Directive(2006/42/EC)
Introduction : Guidelines and general explanations and cautions for preparation
Main subject : A provision for the application of guidelines, legal obligations or authority to the member countries
✧ Annex I : Essential requirements for health and safety regarding design and manufacturing
✧ Annex II : Matters concerning the Declaration of Conformity
✧ Annex III : CE – Marking
✧ Annex IV : List of machinery that must be tested in the EU format by the Notified Body
✧ Annex V : Declaration of Conformity and Technical Files
✧ Annex VI : EU Type Test
✧ Annex VII : Notified Body Criteria
❀ Standard classification
European Standard for machine safety can be divided into A, B, and C standard. The product must meet all 3 applicable standards.
❄ standard A (EN 414, EN 12100)
Standard that define basic concepts, design principles, and general aspects as basic safety standard
❄ Standard B (EN 13850, EN 13849-1, EN 60204-1 and etc.)
Safety-related standard applicable to machines that are widely classified according to their basic use and purpose.
1) Standard B1 for specific safety aspects (Electricity, Safety Distance, Surface Temperature, Noise and etc.)
2) Standard B2 for safety-related devices and mechanical components (emergency switches, pressure sensing devices and etc.)
❄ Standard C
Standard that specify detailed safety requirements for a particular machine or group of machines.
❀ Applied Devices
1. Products with at least one operating part, with electricity, air pressure, hydraulic pressure, weight and etc. as the source of energy
2. Parts of machinery that are involved in health or safety; equipment that is shipped on separateness

❀ CE Machinery Directive(2006/42/EC) core requirement
The documentation requirements that the manufacturer must comply with are in the essential health and safety requirements of Annex I of the MD, documenting whether the machine is being used safely and properly alerting the user if any remaining risks are present
Required requirements as below;
1. the principle of safety, design that facilitates handling, Ergonomics, Control system, Protects against mechanical hazards, electrical and other energy temperatures, other hazards, such as fire and explosion, common to all types of machinery, such as noise emission, Vibration, Radiation and hazardous materials, maintenance and maintenance, Cleaning, Display and Warning of Information
2. Requirements applicable to certain classes of machinery, such as food, cosmetics, and medicines
3. Requirements for offsetting the risk of machine mobility
4. Requirements to offset the risk of lift operations
5. Requirements for underground work machines
6. Requirements for mechanical lift personnel
❀ The certification procedure
There are four methods as follows;
A) High-risk machinery described in Annex 4
Accreditation through the notified Body (NB)
B) General machinery other than Annex 4 (Anex II A)
Acquired by Declaration of self-conformity (with a CE mark as proof of conformity with the finished product)
C) Declaration of self-conformity with respect to Machine Parts (Annes II B)
Acquired by self-compliance declaration (with a CE mark as proof of conformity with the finished product)
D) Declaration of self-conformity with safety components (Annes II C)
Acquired by self-compliance declaration (with a CE mark as proof of conformity with the finished product)
Note: In the case of B) and C), a Declaration of self-conformity can be made and the relevant technical documents stored to declare compliance with the specification, but CE marks cannot be affixed to the product or packaging.
❉ ❉ ❉
IGC’s Competency
- IGC works with European Notified Body (NB) to provide product certification services such as CE Machinery, CE MDD, CE IVD and CE LVD.
- IGC have years of technical skills and expertise, as well as up-to-date knowledge of a range of specific legal requirements in major markets around the world.
- Provide knowledge and services to support your overall global operations.
❉ ❉ ❉
Related Services from IGC
01System certification (ISO 13485, ISO 15378, ISO 14155)
02Product certification (European CE certification, clinical evaluation, medical device registration [Eurasia, China, USA, Thailand, Taiwan])
03Certification of Auditor Qualifications
04Professional manpower training and education
CE PED | Pressure equipment
Introduction to European Product Certification
PED is abbreviation of Pressure Equipment Directive (2014/68/EU), which is a pressure equipment instruction that applies to design, manufacture and conformity assessment of pressure equipment and components with a maximum permissible pressure of 0.5 bar or more. Manufacturers wishing to export their products to EU countries must be certified in accordance with the Pressure Equipment Directive, which was amended from 97/23/EC to 2014/68/EU as of July 19, 2016.
Proof of pressure equipment (CE marking)
Pressure instruments are classified into four stages of Category I, II, III, and IV according to the maximum allowable pressure (PS), fluid use, volume, and external diameter (DN), and are subject to the module requirements applicable to that category as shown in the table below. For detailed technical requirements for conformity assessment, European technical specifications such as EN 1345, AD2000, and ASME codes are applied.
Annex II, Conformity assessment table
Category |
Modules |
I |
Module A |
II |
Modules A2, D1, E1 |
III |
Modules B(Design type)+D, B(Design type)+F, B(Production type)+E, B(Production type)+C2, H |
IV |
Modules B(Production type)+D, B(Production type)+F, G, H1 |
Annex III, Conformity assessment table
Module A |
Internal production management |
Module A2 |
Internal production management + Product inspection under supervision at random cycles |
Module B |
EU-Type inspection (Production type)
EU-Type inspection (Design type) |
Module C2 |
Formal conformity based on internal production management + Product inspection under supervision at random cycles |
Module D/D1 |
D: Formal suitability based on quality assurance of production process
D1: Quality Assurance of Production Process |
Module E/E1 |
E: Formal suitability based on product quality assurance
E1: Quality Assurance of Inspection and Testing of Final Products |
Module F |
Formal suitability based on product verification |
Module G |
Conformity based on unit verification |
Module H/H1 |
H: Conformity based on full quality assurance
H1: Conformity based on complete quality assurance + Design inspection |
❉ ❉ ❉
IGC’s Competency
♕ IGC provides product certification services to companies that want to export their products to Europe based on years of experience.
♕ We provide the most competitive product certification service through cooperation with leading overseas product certification institutions.
♕ ISO 9000, Companies that already comply quality management systems can make the most of their current quality systems and meet the essential safety requirements of PEDs.
♕ If the manufacturer has not been formally certified as a Quality Management System, IGC will provide certification authority services in accordance with the PED QA approach.
❉ ❉ ❉
Related Services from IGC
01System certification (ISO 13485, ISO 15378, ISO 14155)
02Product certification (European CE certification, clinical evaluation, medical device registration [Eurasia, China, USA, Thailand, Taiwan])
03Certification of Auditor Qualifications
04Professional manpower training and education
CE SPVD | Simple pressure container Introduction to European Product Certification
SPVD is a Simple Pressure Vessel Directive that is a weld container with an internal pressure of 0.5 or higher, applies to pressure vessels that contain air or nitrogen and target all non-flammable fluids, and is manufactured continuously.
Manufacturers who wish to export their products to EU countries shall be certified according to the simple pressure container guidelines, and the simple pressure container guidelines have been revised to 2014/29/EU as of April 20, 2016
Applied scope/ Excluded scope
1. Applied scope
All pressure vessels containing air or nitrogen and non-flammable as a welding vessel with an internal pressure of 0.5 bar or higher.
⚜ Areas and assemblies that affect the strength of the vessel under pressure must be made of non-alloyed steel, non-alloyed aluminum, or sturdy aluminum alloy that does not undergo aging.
⚜ The container consists of a cylindrical part or cylindrical part of a circular cross-sectional area closed by a stool on the outside, and a plate rotating coaxially or a stool rotating coaxially.
⚜ The maximum operating pressure of the container must not exceed 30 bar, and the product of the inner volume and pressure of the container must not exceed 10,000 bar/liter.
⚜ The minimum operating temperature of the container should not be lower than -50℃ and the maximum operating temperature should not be higher than 300℃ for steel and 100℃ for aluminum and aluminum alloy containers.
2. Excluded scope
⚜ Container designed to prevent radioactive leakage for use in nuclear power
⚜ Container used for propulsion devices or equipment of ships or aircraft
⚜ Fire extinguisher
❉ ❉ ❉
Essential requirements
The prerequisites for SPVD are below:
1) Vessels with a maximum permissible pressure (PS) x capacity (V) exceeding 50 bar.L must meet the essential safety requirements of Annex I
2) Vessels with a maximum permissible pressure (PS) x capacity (V) of 50 bar.L or less must be designed and manufactured according to the engineering practice of one of the member states.
❉ ❉ ❉
The importance of SPVD
❉ ❉ ❉
Conformity assessment procedure
For simple pressure vessels, the module is determined according to the maximum allowable pressure (PS) x capacity (V).
Module B + |
Module C1 |
EU- Type inspection + Product testing under formal conformity and supervision based on internal production management |
Module C2 |
EU- Type inspection + Product testing under formal conformity and supervision based on internal production management at random cycles |
Module C3 |
EU- Type inspection + Formal compatibility based on internal production management |
❉ ❉ ❉
IGC’s Competency
♔ IGC provides certification services for simple pressure vessels in agreement with FQC (First Quality Certification, NB2674), a European accreditation body.
♔ The auditors of IGC contribute to the continuous development of customers by accurately evaluating the suitability of the management system through the technology and expertise accumulated over the years.
♔ As a reliable global leader in product certification, IGC provides certifications for ISO SPVD as well as PED and hot-water boilers.
♔ IGC has up-to-date knowledge of a wide range of specific scopes and legal requirements in major markets around the world.
♔ IGC provides knowledge and services to support your entire global operations.
Related Services from IGC
01System certification (ISO 13485, ISO 15378, ISO 14155)
02Product certification (European CE certification, clinical evaluation, medical device registration [Eurasia, China, USA, Thailand, Taiwan])
03Certification of Auditor Qualifications
04Professional manpower training and education
Hot-water boiler
Introduction to European Product Certification
92/42/EEC is a directive on the efficiency requirements for new hot water boilers burning on liquid or gaseous fuels rated at 4 kW ~ 400 kW and is part of the European Union's SAVE program on promoting energy efficiency.
The purpose of this directive is to improve energy efficiency by setting efficiency requirements for hot water boilers that burn as liquid or gas.
Scope
Hot water boilers that use liquid or gaseous fuel with a rated output of 4 Kw ~ 400 Kw (standard boiler, low temperature boiler, gas condensing boiler)
❉ ❉ ❉
Excluded Scope
⚙ Hot water boiler that can be burned by various fuels including solid fuel
⚙ Hot water preparation equipment
⚙ Boiler designed to be burned by fuels(Industrial waste gas, biogas, etc.) with properties different from those of liquid and gaseous fuels on the market.
⚙ Cooking utensils and appliances installed in the building to provide central heating and sanitary hot water with the main purpose of heating
⚙ Equipment designed for the production of stored sanitary hot water with a rated output of less than 6kW using gravity circulation
⚙ Single use Boiler
❉ ❉ ❉
Efficiency requirements
⛭ Rated power: Operates at an average boiler water temperature of 70 °C at rated power (Pn) expressed in kW
⛭ Partial load: Operates with a load of 30% at the average water temperature of the boiler depending on the type of boiler
Type of boiler |
Output range |
Efficiency at rated power |
Efficiency at partial load |
|
Kw |
Average boiler water temperature (in ℃) |
Efficiency requirement (in %) |
Average boiler water temperature (in ℃) |
Efficiency requirement (in %) |
Standard boiler |
4 - 400 |
70 |
≥ 84 + 2
logPn |
≥ 50 |
≥ 80 + 3 |
Low temperature boiler + |
4 - 400 |
70 |
≥ 87.5 + 1.5
logPn |
40 |
≥ 87.5 + 1.5 |
Gas condensing boiler |
4 - 400 |
70 |
≥91 + 1
logPn |
30(**) |
≥ 97 + 1
logPn |
(*) Condensing boilers using liquid fuel included
(**) Water temperature of Boiler
❉ ❉ ❉
Conformity assessment procedure
Module B + |
Module C |
EU- Type Inspection + Type suitability |
Module D |
EU- Type Inspection + Production quality assurance |
Module E |
EU- Type Inspection + Production quality assurance |
❉ ❉ ❉
IGC’s Competency
⛯ IGC has been accredited by IAS, an accreditation body in the United States, and provides certification services for management systems.
⛯ The auditors of IGC contribute to the continuous development of client by accurately evaluating the suitability of the management system through the technology and expertise accumulated over the years.
⛯ IGC is a trusted global leader in management system certification, providing certification for quality, environment and other management systems.
⛯ IGC has up-to-date knowledge of a wide range of specific scope and legal requirements in major markets around the world.
⛯ IGC provides knowledge and services to support your entire global operation.
❉ ❉ ❉
Related Services from IGC
01System certification (ISO 13485, ISO 15378, ISO 14155)
02Product certification (European CE certification, clinical evaluation, medical device registration [Eurasia, China, USA, Thailand, Taiwan])
03Certification of Auditor Qualifications
04Professional manpower training and education
Gas Appliance | Gas fuel combustion equipment
Introduction to European Product Certification
Appliances burning gaseous fuels (EU 2016/426) introduction
EU 2016/426 is a European gas appliance regulation that applies to gas appliances and accessories designed for hot water production, refrigeration, lighting, heating, washing and cooking purposes.
The European Gas Appliances Directive (GAD 2009/142/EC) issued in March 2017 was replaced by the Gas Appliances Regulation (GAR 2016/426) has been updated and applied from April 2018.
Scope
Gas-fired cooking, heating, hot water, refrigeration, and laundry equipment are included, and generally used household and commercial products excluding industrial products.
❉ ❉ ❉
Excluded scope
⁘ Products used in industrial processes carried out in industrial sites
⁘ Products used in aircraft and railroads
⁘ Research purpose products for temporary use in the laboratory
❉ ❉ ❉
Conformity assessment procedure
연In the case of products that are continuously produced, the manufacturer must evaluate the suitability by combining one of Modules C2, D, E, and F according to Module B-Production Type.
Module B
(Production Type)
+ |
Module C2 |
EU-Type Inspection + Formal conformity based on internal production management and product inspection under supervision at random cycles |
Module D |
EU-Type Inspection + Form conformity based on quality assurance of the production process |
Module E |
EU-Type Inspection + Form conformity based on product quality assurance |
Module F |
EU-Type Inspection + Form conformity based on product verification |
In the case of products produced in single units or in small quantities, the manufacturer can also be evaluated for conformity with Module G (Unit Verification).
❉ ❉ ❉
Related Services from IGC
01System certification (ISO 13485, ISO 15378, ISO 14155)
02Product certification (European CE certification, clinical evaluation, medical device registration [Eurasia, China, USA, Thailand, Taiwan])
03Certification of Auditor Qualifications
04Professional manpower training and education
E-MARK Certification
Introduction of European Product Certification
E-Mark Certification Overview
EU vehicle certification is a comprehensive conformity regulation of automobiles, including automobiles, related parts and electronic equipment, of the Economic Commission for Europe (ECE) and the European Union (EU).
Companies wishing to sell automobiles and auto parts to the European Union market must obtain the E-mark certification, an approval scheme for the EU automotive sector, from the Ministry of Transport of the EU. Only automobiles or auto parts with E-mark certification can be exported and traded freely within the European Union.
Applicable Items
In general, cars, trucks, buses, trailers and other road-driving vehicles (including vehicle-mounted ESA/parts) are now required to comply with UNECE Regulation 10 for electromagnetic compatibility. Accordingly, UNECE Regulation 10 for system EMC certified for type approval is marked with a capital “E” E-Mark.
Items subject to e-Mark:
Vehicles and parts complying with EU directives: steering system, brakes, various lamps, heating systems, speedometers, helmets, child seats, exhaust pipes, anti-theft devices, tires, seat belts, connecting devices, automotive glass, cooling systems, storage protection, interior material of bus, horn, seat and headdress, etc.
❉ ❉ ❉
Applied Standard
UN-ECE Regulation 10 that replaced the ‘Automotive Electromagnetic Compatibility Directive’ 2004/104/EC (AEMCD) in November 2014.
Items subject to e-Mark:
2007/46/EC: approval of motor vehicles and their trailers, and of systems, components and separate technical units intended for such vehicles
❉ ❉ ❉
Difference between E-Mark and e-Mark
E-Mark
E-Mark is the UN mark for approved vehicles and vehicle parts sold to the EU, and is a certification for type approval of parts or systems of automobiles.
The E-Mark means conformity to the world standard ECE Regulation.
e-Mark
The e-Mark is an EU mark for approved vehicles and vehicle parts sold to the EU, and is a certification that includes type approval for all vehicles as well as parts and systems of automobiles.
The e-Mark means compliance with the European Automotive EMC Directive, which is a mandatory European directive.
❉ ❉ ❉
Certification Mark
E-Mark European Economic Commission (ECE, UN ministry) Certification, E-Mark
 E-Mark Certification Mark
E-Mark Certification Mark
❖ The E-Mark is a regulation issued by the European Economic Commission.
❖ The E-Mark logo is a circular frame.
❖ The number is the EU country or state number that has been granted approval. If approved by one country/state, it is considered sufficient for all other member states.
❖ The country/state codes are as follows:
E1—Germany |
E16—Norway |
E32—Latvia |
E46—Ukraine |
E2—France |
E17—Finland |
E33— |
E47—South Africa |
E3—Italy |
E18—Denmark |
E34—Bulgaria |
E48—New Zealand |
E4—Netherlands |
E19—Romania |
E35— |
E49—Cyprus |
E5—Sweden |
E20—Poland |
E36—Lithuania |
E50—Malta |
E6—Belgium |
E21—Portugal |
E37—Turkey |
E51—Korea |
E7—Hungary |
E22—Russia |
E38— |
E52—Malaysia |
E8—Czech Republic |
E23—Greece |
E39—Azerbaijan |
E53—Thailand |
E9—Spain |
E25—Croatia |
E40—Macedonia |
E54— |
E10—Yugoslavia |
E26—Slovenia |
E41— |
E55— |
E11—United Kingdom |
E27—Slovakia |
E42—European Community |
E56—Montenegro |
E12—Austria |
E28—Belarus |
E43—Japan |
E56—Tunisia |
E13—Luxembourg |
E29—Estonia |
E44— |
|
E14—Switzerland |
E31—Bosnia |
E45—Australia |
|
EU Certification (EC), e-Mark
 EU(EC), e-Mark
EU(EC), e-Mark
❖ The e-Mark is a safety certification mark based on EU directives.
❖ The EU Commission requires Member States to apply it to automobiles, parts and systems.
❖ The e-Mark logo is a rectangular frame.
❖ The number is the EU country or state number that has been granted approval. If approved by one country/state, it is considered sufficient for all other member states.
❖ The country/state codes are as follows:
e1 – Germany |
e11 – United Kingdom |
e24 – Ireland |
e2 – France |
e12 – Austria |
e26 – Slovenia |
e3 – Italy |
e13 – Luxembourg |
e27 – Slovakia |
e4 – Netherlands |
e17 – Finland |
e29 – Estonia |
e5 – Sweden |
e18 – Denmark |
e32 – Latvia |
e6 – Belgium |
e19 – Romania |
e34 – Bulgaria |
e7 – Hungary |
e20 – Poland |
e36 – Lithuania |
e8 – Czech Republic |
e21 – Portugal |
e49 – South Cyprus |
e9 – Spain |
e23 – Greece |
e50 – Malta |
❉ ❉ ❉
Test Standard
Regulation 10 Concerning the approval of vehicles with regard to electromagnetic compatibility
| Testing Equipment |
Test Items |
Test Standard |
| EMI Measurement System |
Radiated Emission |
Regulation No.10, CISPR 25 |
| Conducted Emission (Current) |
| Conducted Emission (Voltage) |
| EMS Test System |
RI Test (ALSE) |
Regulation No.10,ISO 11452-2 |
| BCI Test |
Regulation No.10,ISO 11452-4 |
| Transient |
Transient Emission |
Regulation No.10, ISO 7637-2 |
| Transient Immunity |
Regulation No.10, ISO 7637-2 |
❉ ❉ ❉
Certification Process
1. Application
- Fill out and submit the application
2. Documents & Samples
- Preparation of technical documents and samples
3. Conduct the Test
- Conduct the sample test
4. Conformity of Production
- Conduct on-site inspection to evaluate the ability to produce the product according to the technical standards of the production plant
*Manufacturers who have obtained ISO9000 and other quality system certificates issued by EU national certification bodies may be exempt from factory inspection.
5. Test Report
- Issuance of test report, submitting with other technical documents to the Ministry of Transport of European countries
6. Certificate Issuance
- Comprehensive review of the document review and the test results for the relevant guidelines and the on-site audit results for the manufacturing plant, and if passed, a certificate is issued by the European Ministry of Transport.
7. Regular Factory Inspection
- The issuing authority may track and inspect factory inspections of certified product manufacturers on a regular or irregular basis.
*For factories that have obtained ISO9001, the factory inspection period may be shortened or exempted in some cases.
❉ ❉ ❉
Preparation Item
Submitted documents must be written and submitted in English.
1. Manufacturer-related information
- - Manufacturer’s name
- - Manufacturer's address
- - Copy of Business Registration Certificate
2. Product-related information
- - Product Manual/User Manual
- - Product Technical Drawing
- - Parts list
- - Parts Layout Drawing
- - Electric block diagram
- - Product photo
- - Circuit Diagram
3. Manufacturing plant ISO 9001 or IATF 16949 certificate
❉ ❉ ❉
IGC’s Competency
⁙ IGC has up-to-date knowledge of a wide range of specific scope and legal requirements in major markets around the world, and provides the knowledge and services to support your entire global operation.
⁙ IGC provides professional and competitive services to customers in the automotive sector.
⁙ IGC provides E-Mark certification services in cooperation with Croatian transport ministry in Europe.
❉ ❉ ❉
Related Services from IGC
01System certification (ISO 13485, ISO 15378, ISO 14155)
02Product certification (European CE certification, clinical evaluation, medical device registration [Eurasia, China, USA, Thailand, Taiwan])
03Certification of Auditor Qualifications
04Professional manpower training and education
